Track types
Different track types support different types of data (figure 28.2). Key examples are:
- Sequence Track (
 )
) A Sequence Track contains one or more sequences. It is usually used for the reference sequences of a genome or for sets of contigs.
- Reads Track (
 )
) A Reads Track contains a read mapping, i.e. reads aligned against a set of reference sequences.
- Variant Track (
 )
) A Variant Track contains information about various types of variants. These can include SNVs, MNVs, replacements, insertions and deletions, along with details about each variant, for example the allele, its length and frequency.
- Annotation Track (
 )
) An Annotation Track contains information about a certain type of annotation, for example gene annotations, mRNA annotations, or a set of target regions. Some analyses require specific types of Annotation Tracks as input. For example, gene tracks and mRNA tracks are commonly used when running RNA-Seq Analysis.
- Coverage Graph Track (
 )
) A Coverage Graph Track contains a graphical display of the coverage at each position of a reads track. It can be produced by Graph tools.
- Expression Track (
 )
) An Expression Track contains expression values for genes or for transcripts. See Expression tracks for further details.
- Statistical Comparison Track (
 )
) A Statistical Comparison Track contains results from a differential expression analysis. See Output of the Differential Expression tools for further details.
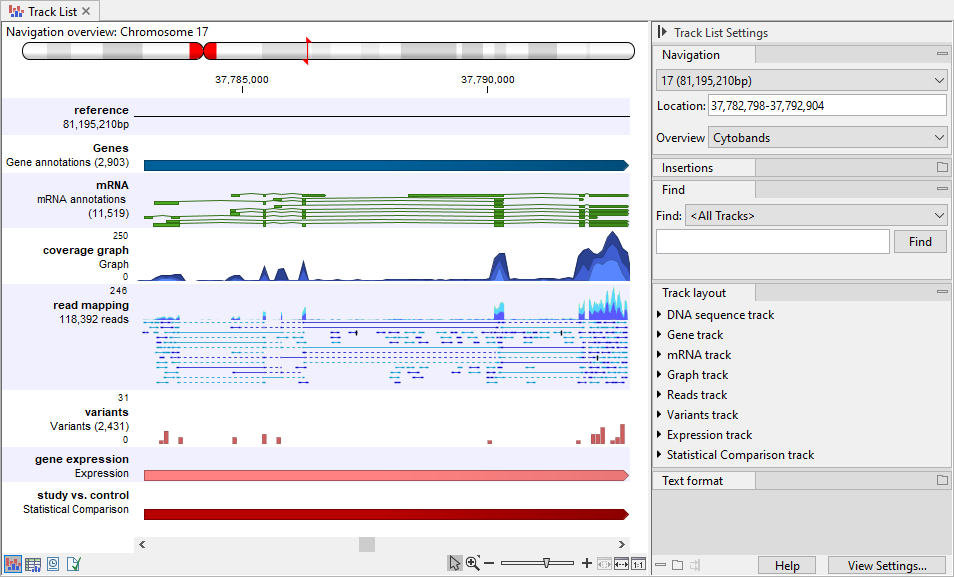
Figure 28.2: A track list containing different types of tracks. From the top: a Sequence Track, two Annotation Tracks containing genes and mRNAs, respectively, a Coverage Graph Track, a Reads Track, a Variant Track, an Expression Track, and a Statistical Comparison Track.