Running workflows in batch mode
Running analyses in batches occurs when:
- The Batch checkbox at the bottom of input steps in the launch wizard has been checked, and/or
- The workflow contains one or more Iterate control flow elements. Steps downstream of Iterate elements and upstream of Collect and Distribute elements, if present, are run once for each batch unit (see Iterate and Collect and Distribute elements).
A batch unit consists of the data that should be analyzed together. The grouping of data into batch units is defined after the inputs for analysis have been selected.
Defining batch units based on the organization of the input data
For simple workflows, batch units can be defined based on how the input elements or files are organized. This is identical to defining batch units when launching a tool, as described in Running the analysis and organizing the results.
Here "simple workflows" means workflows with just one analysis input that changes per batch, for example, the sets of sequencing reads to be mapped, where the same reference sequence is used for every mapping. This equates to a workflow without any Iterate elements being run in Batch mode, or a workflow with just one Iterate element being run (not in Batch mode).
Defining batch units based on metadata
When launching any workflow, batch units can be defined using metadata. For more complex scenarios, this will be the only option. Such scenarios include:
- Where there is more than one level of batch units. This could be:
- A workflow with more than one Input element, where the inputs to both of these should be grouped into batch units. An example of such a workflow is described in Advanced workflow batching.
- A workflow containing more than one Iterate element.
- A workflow containing containing an Iterate element that will be run in Batch mode. An example of this is described in the "RNA-Seq and differential gene expression analysis" tutorial, available from https://resources.qiagenbioinformatics.com/tutorials/RNASeq-DGE-analysis.pdf.
- Where Iterate or Collect and Distribute elements in the workflow have been configured to require metadata.
Note: When launching a workflow containing analysis steps that require metadata, the metadata provided to define batch units is also used for those analysis steps. For example, in the RNA-Seq and Differential Gene Expression Analysis template workflow, metadata provided to define batch units is also used for the Differential Expression for RNA-Seq step.
There are two ways metadata defining batch units can be provided:
- Using a CLC Metadata Table In this case, the data elements selected as inputs must already have associations to this CLC Metadata Table.
If a CLC Metadata Table with data associated to it has been selected in the "Select Workflow Input" step of a workflow, that table will be pre-selected in the "Configure batching" step of the launch wizard. You can specify the column that batch units will be based on. Data associated with the table rows for each unique value in that column make up the contents of the batch units. The contents can be refined using the fields below the preview pane (figure 15.74).
Outputs from the workflow that can be unambiguously identified with a single row of the CLC Metadata Table will have an association to that row added. Outputs derived from two or more inputs with different metadata associations will not have associations to this CLC Metadata Table.
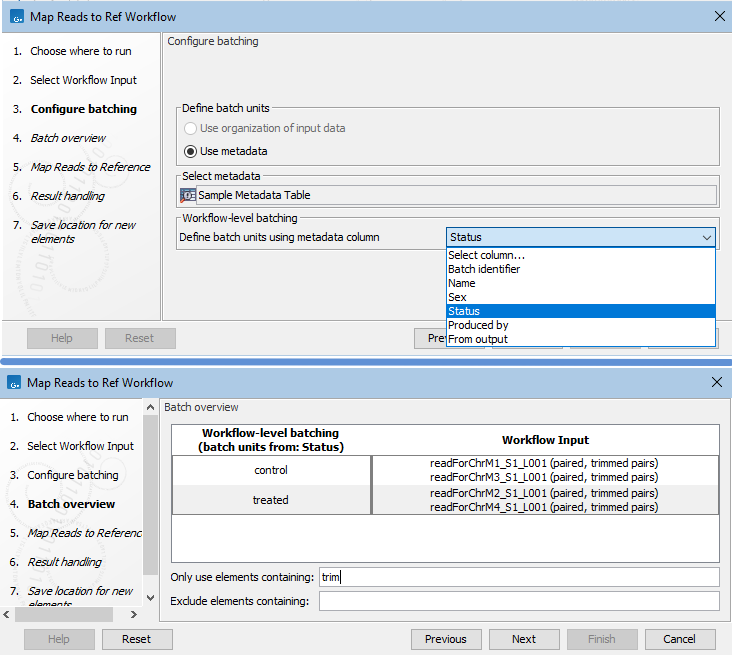
Figure 15.74: A CLC Metadata Table with data associated to it was selected as input to a workflow being launched in Batch mode. In the Configure batching wizard step, the metadata source is pre-configured. The column to base batch units on can be selected (top). The Batch overview step shows the data elements in each batch unit. Here "trim" has been entered in the "Only use elements containing" field, so only elements containing the term "trim" in their names are included in the batch units (bottom). - Using an Excel, CSV or TSV format file. The metadata in the file is imported into the CLC software at the start of the workflow run. Requirements for this file are:
- The first row must contain column headers.
- The first column must contain either the exact names of the files selected or at least enough of the first part of the name to uniquely identify each file with the relevant row of the metadata file. If data is being imported on-the-fly, the file name can include file extensions, but not the path to the data.
- A column containing information that defines how the data should be grouped for the analysis, i.e. the information that defines the batch units. In many cases, this column contains sample identifiers. This may be the first column if there are as many batch units as input files.
When the data being imported is paired sequence reads, the first column would contain the names of each input file, and another column would uniquely identify each pair of files (figure 15.75).
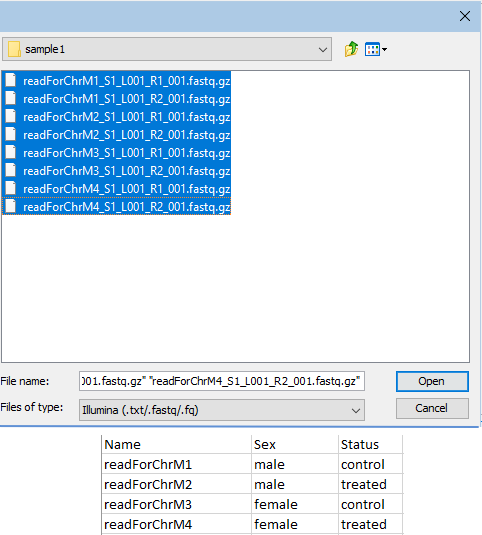
Figure 15.75: Paired fastq files from two samples were selected for import (top). The Excel file with information about this data set contains a header row and 4 rows of information, one row per input file. The contents of the first column contain enough of each file name to uniquely identify each input file. The second column contains sample IDs.
If there is a tool in the workflow that requires descriptive information, for example, factors for statistical testing in Differential Expression for RNA-Seq, then the file should also contain columns with this information.
For example, if a data element selected in the Navigation Area has the name
Tumor_SRR719299_1 (paired) (Reads), then the first column could contain that name in full, or just enough of the first part of the name to uniquely identify it. This could be, for example,Tumor_SRR719299. Similarly, if a data file selected for on-the-fly import is at:C:\Users\username\My Data\Tumor_SRR719299_1.fastq, the first column of the Excel spreadsheet could containTumor_SRR719299_1.fastq, or a prefix long enough to uniquely identify the file, e.g.Tumor_SRR719299.
Example: On-the-fly import of single end reads based on metadata
In figure 15.76, a workflow with a single input is being launched in batch mode. The eight files selected contain Illumina single end reads. This raw data will be imported on the fly using metadata to define the batch units. The metadata column in the Excel file that contains information defining the batch units has been specified. Here, files with the same value in the SRR_ID column will be imported and analyzed together.
Each row in the SRR_ID column has a unique entry, so 8 batch units will be made, with one sequence file in each batch unit. If a column containing fewer unique values was selected, one or more batch units would consist of several files. This is illustrated in figure 15.76.
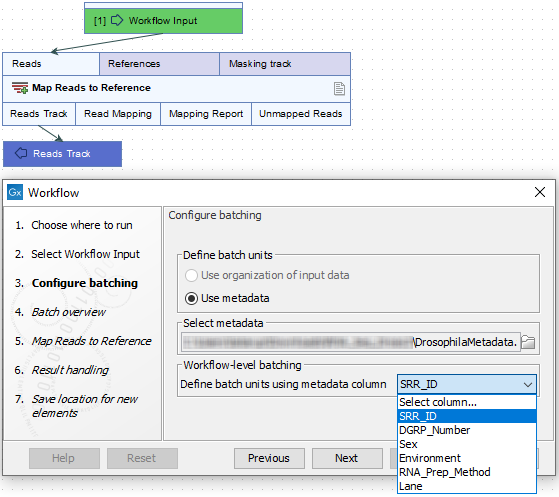
Figure 15.76: Batch units are defined according to the values in the SRR_ID column of the Excel file that was selected.
In the next step, a preview of the batch units is shown. The workflow will be run once for each entry in the left hand column, with the input data grouped as shown in the right hand column (figure 15.77).
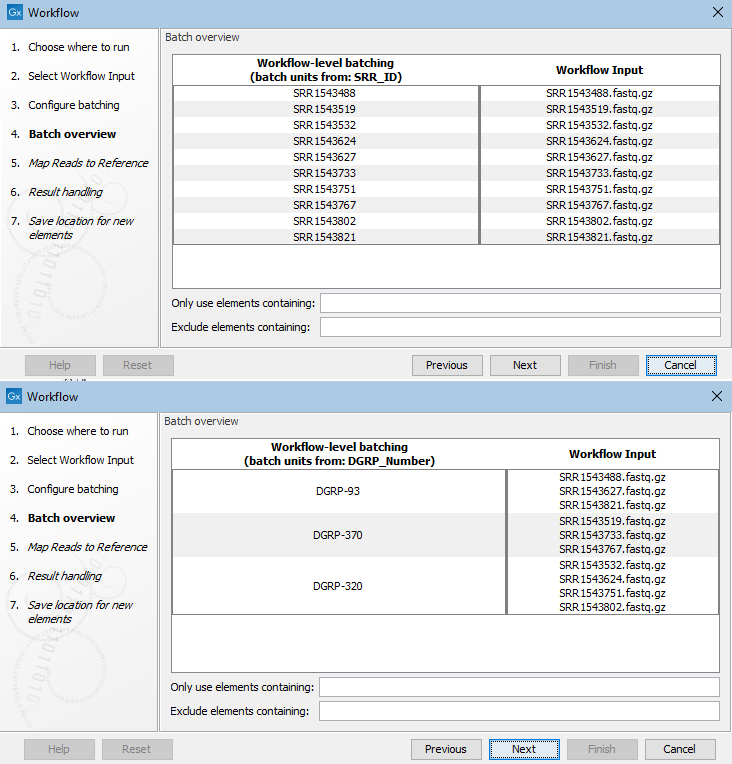
Figure 15.77: The Batch overview step allows you to review the batch units. In the top image, a column called SRR_ID had been selected, resulting in 8 batch units, so 8 workflow runs, with the data from one input file to be used in each batch. In the lower image, a different column was selected to define the batch units. There, the workflow would be run 3 times with the input data grouped as shown.
Example: On-the-fly import of paired end reads based on metadata
When importing data on-the-fly and organizing batch units based on metadata, the metadata must have a row per file being imported. For paired data, this means at least 2 rows per sample. One column in the file must contain information about which files belong together. This allows the sequence list created to be associated with the relevant information.
The contents of an Excel file with information about 2 sets of paired files containing Sanger data are shown in figure 15.78.
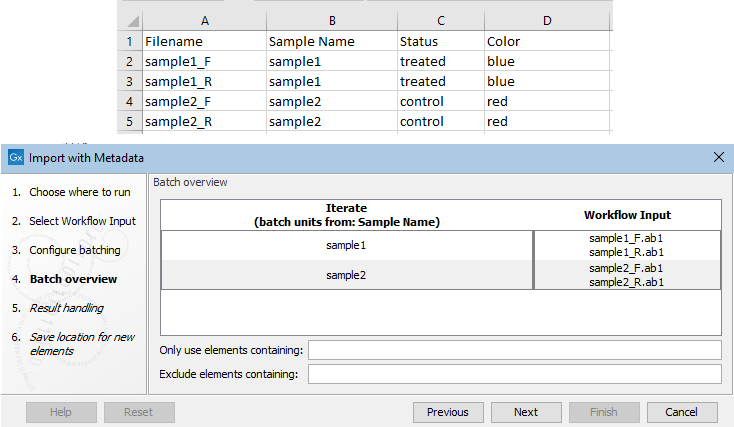
Figure 15.78: An Excel file at the top describes 4 Sanger files that make up two pairs of reads. The "Sample Name" column was identified as the one indicating the group the file belongs to. Information about the relevant sample appears in each row. At the Batch overview step, shown at the bottom, you can check the batch units are as intended.
Each row for data that is in the same batch unit must contain the same descriptive information. Where there is conflicting information for a given batch unit, the value for that column will be ignored. If all entries for a given column are conflicting, the column will not appear in the resulting CLC Metadata Table.
Saving results from workflows run in batch mode
When a workflow is run in batch mode, options are presented in the last step of the wizard for specifying where to save results of individual batches (see Running the analysis and organizing the results).
If the workflow contains Export elements, an additional option is presented, Export to separate directories per batch unit (figure 15.79). When that option is checked, files exported are placed into separate subfolders under the export folder selected for each export step.
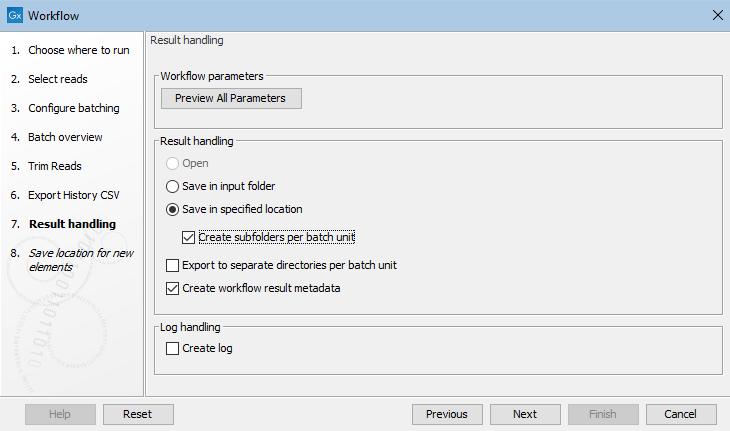
Figure 15.79: Options are presented in the final wizard step for configuring where outputs and exported files from each batch run should be saved.