RNA-Seq and Differential Gene Expression Analysis workflow
The RNA-Seq and Differential Gene Expression Analysis workflow calculates gene expression profiles per sample, and then performs differential expression analysis across samples. It also generates various reports and visualizations of the expression profiles and differential expression results. The ability to run parts of the workflow on a per-sample basis and other parts based on all samples, is possible due to the Iterate and Collect and Distribute workflow elements, see Workflow control flow elements.
Validation of results should be carried out. Some common workflow customizations are provided at the end of this section.
Inputs to the workflow
To run this workflow, you will need:
- Trimmed reads Reads can be trimmed using the Trim Reads tool (Trim Reads) or the Prepare Raw Data template workflow (Prepare Raw Data).
- Metadata containing information about the samples. This can be an Excel, CSV or TSV format file (figure 15.103), or a CLC Metadata Table. The metadata provided should include the factors relevant for differential expression analysis (e.g. treatment level, sex, etc.). For details about providing metadata when launching a workflow, see Defining batch units based on metadata.
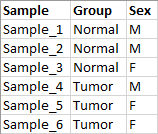
Figure 15.103: Metadata describing samples from a tumor-normal comparison experiment.
Launching the workflow
The RNA-Seq and Differential Gene Expression Analysis template workflow is at:
Workflows | Template Workflows | Basic Workflow Designs (![]() )
| RNA-Seq and Differential Gene Expression Analysis (
)
| RNA-Seq and Differential Gene Expression Analysis (![]() )
)
Launch the workflow and step through the wizard.
- Select the trimmed reads to be processed.
- In the next steps, select the reference sequence, genes, mRNA, and CDS tracks, and finally, the gene ontology. See the customizations at the end of this section if not all inputs are relevant for your species.
- Next, choose "Use metadata" for defining the batch units. Select the CLC Metadata Table or the Excel, CSV or TSV format file containing information about the samples, and choose the column used for grouping the reads into batch units (figure 15.104). For further details see Defining batch units based on metadata.
- In the next step, you can review the batch units resulting from your selections above.
- You can configure next the strand specificity and library type used to produce your data.
- Specify next the differential expression settings (figure 15.105).
- In the next step, you can click on Preview All Parameters to review your settings.
- In the final step, choose a location to save the results to.
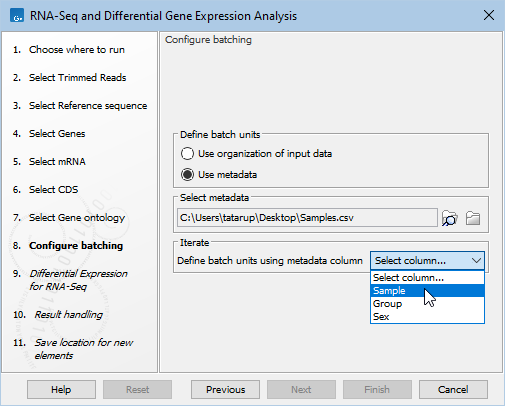
Figure 15.104: After selecting the metadata source, specify the column containing the information that groups the reads appropriately for the RNA-Seq analysis. Usually this would be a column containing a unique identifier per sample.
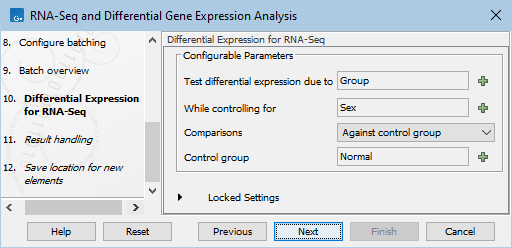
Figure 15.105: Specify the settings for the differential expression analysis. The columns from the metadata provided earlier will be available for selection in relevant options.
Tools in the workflow and generated outputs
The RNA-Seq and Differential Gene Expression Analysis workflow contains several tools and produces multiple outputs. The workflow has been configured to save many of the outputs to subfolders. These are created automatically within the folder that you selected to save results to when launching the workflow. See Configuring custom output names for details.
The following tools produce elements per sample:
- QC for Sequencing Reads outputs one report that is useful for validating the quality of the reads after trimming. It is saved to the subfolder QC & Reports/<Batch unit>.
- RNA-Seq Analysis outputs:
- One Gene Expression Track containing the gene expression profile. It is saved to the subfolder Gene Expression Tracks.
- One report summarizing the RNA-Seq analysis results. It is saved to the subfolder QC & Reports/<Batch unit>.
- Create Sample Report generates one report summarizing the QC for Sequencing Reads and RNA-Seq Analysis reports for that sample. The sample report is not saved as output. It is provided as input to the Combine Reports tool, in a downstream step of the workflow.
The following tools output elements across all samples. Their outputs are saved to the subfolder Expression Analysis:
- Differential Expression for RNA-Seq outputs Statistical Comparison Tracks containing the results of the performed tests. The number of output tracks depends on the settings used for the differential expression analysis (figure 15.105).
- Gene Set Test outputs a Gene Ontology enrichment analysis for each Statistical Comparison Track.
- Create Expression Browser outputs a single table containing all Gene Expression Tracks and Statistical Comparison Tracks.
- Create Venn Diagram for RNA-Seq outputs a Venn diagram comparing the overlap of differentially expressed genes from the Statistical Comparison Tracks.
- PCA for RNA-Seq outputs a plot containing the projection of the Gene Expression Tracks into two and three dimensions.
- Create Feature Level Heat Map for RNA-Seq outputs a heat map of the most variable genes across samples.
- Create Sample Level Heat Map for RNA-Seq outputs a heat map of the distance between samples, based on their gene expression profiles.
The following tools output elements across all samples. Their outputs are saved to the folder selected to save results to when launching the workflow:
- Combine Reports outputs one report. It takes the individual sample reports generated by Create Sample Report and generates a single report, useful for comparing the individual sample results.
- Create Track List outputs a track list containing the reference sequence, genes, mRNA, CDS, and the Statistical Comparison Tracks.
Customizing the workflow
Template workflows can be easily edited to add, remove or change analysis steps. See Template workflows for information about how to open a copy of a template workflow for editing.
- RNA-Seq Analysis. The expression profile is at gene level and hence the differential expression is also reported at gene level. If you want to quantify transcript expression instead, use the "Transcript Expression Track" output instead of the "Gene Expression Track" output from the "RNA-Seq Analysis" workflow element.
- Genes track and mRNA track. RNA-Seq Analysis is configured to use of both genes and mRNA tracks. They are also added as inputs to the "Create Track List" workflow element. The "RNA-Seq Analysis" workflow element can be configured with different "Reference types" and any input workflow elements that are not needed can be removed.
- CDS track. A CDS track is added as input to the "Create Track List" workflow element. If you do not have CDS track for your reference genome, remove the "CDS" input workflow element.
- Differential Expression for RNA-Seq. The workflow element is configured for whole transcriptome RNA-Seq data. Options are available for other types of RNA data.
- Gene Set Test. The workflow element requires a GOA database. If you do not have this for your species, remove the "Gene Set Test" and "Gene ontology" workflow elements.
- Create Feature Level Heat Map for RNA-Seq. The workflow element is configured to use the 25 genes that are most variable. Options are available for choosing other genes.