Reads tracks
Reads tracks can be included in a track list with other tracks that are based on a compatible genome, allowing for convenient, comparative visual analysis. For example, the track list shown in figure 31.20 contains, from top to bottom, a cytoband overview, a reads track, an annotation track of target regions, and a variant track.
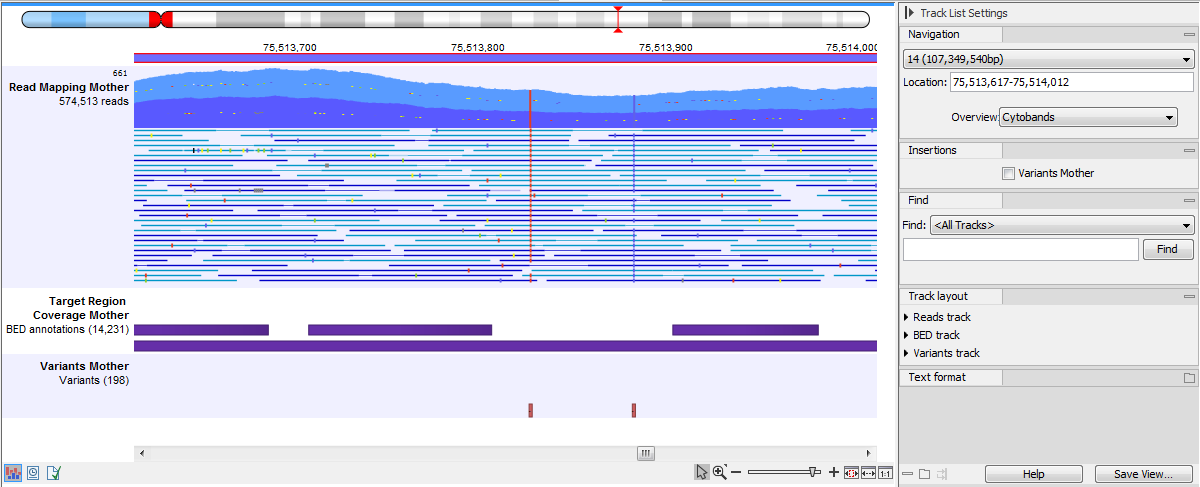
Figure 31.20: A track list containing a reads track, an annotation track and a variant track.
Reads tracks contain only the reads, placed where they mapped using the relevant reference genome coordinates. The information available when viewing a reads track depends on how far you zoom out or in.
Zooming out to an aggregated view
When zoomed out to the point where the mapped reads are aggregated, as shown in figure 31.21, the height of the graph reflects the read coverage of that region, with the shade of blue representing the maximum, average and minimum coverage of the positions in that region:
- Dark blue: maximum read coverage
- Royal blue: average read coverage
- Light blue: minimum read coverage
A tooltip is shown when hovering the mouse cursor over any position in the track, reporting these values and the length of the region where those values apply.
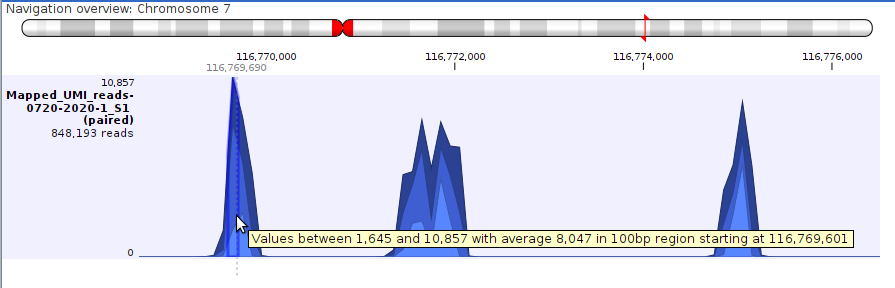
Figure 31.21: Shades of blue in an aggregated reads track represent the maximum, average and minimum reads coverage. Hovering the mouse cursor over a position brings up a tooltip with informaton about the coverage in that region.
Zooming in to a detailed view
When zoomed in fully, the individual nucleotide bases of each read are displayed (figure 31.22). A few reads are shown in detail, with the rest summarized in an coverage graph above. To see more reads in detail, increase the height of the reads track. For mappings where the coverage is deeper than the track height, a vertical scrollbar will appear when hovering the mouse cursor over the right hand side of the reads track. Use this to scroll through and view the details of all reads in the reads track.
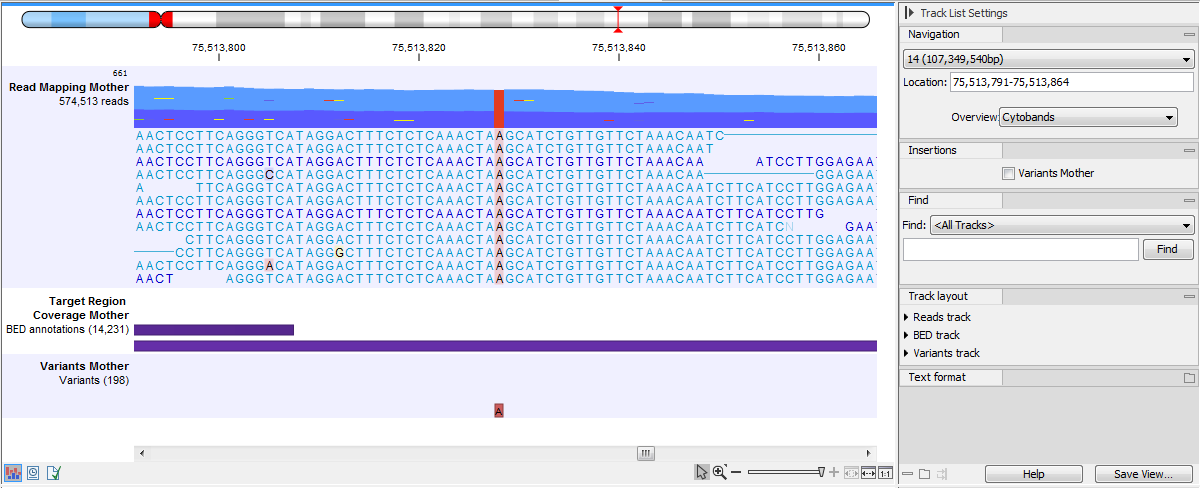
Figure 31.22: Zoom in fully to see the nucleotide bases of each read.
Reads that map across the origin of circular genomes are displayed at both the start and end of the mapping, and are marked with double arrows » at the ends of the read to indicate that the read continues at the other end of the reference sequence.
When fully zoomed into a reads track, you can:
- Place the mouse cursor on a particular read and right-click to reveal a menu. Choose the option Selected Read to open a submenu where there are options for copying the reads, opening it in a new view, or using it as a query in a BLAST search (figure 31.23).
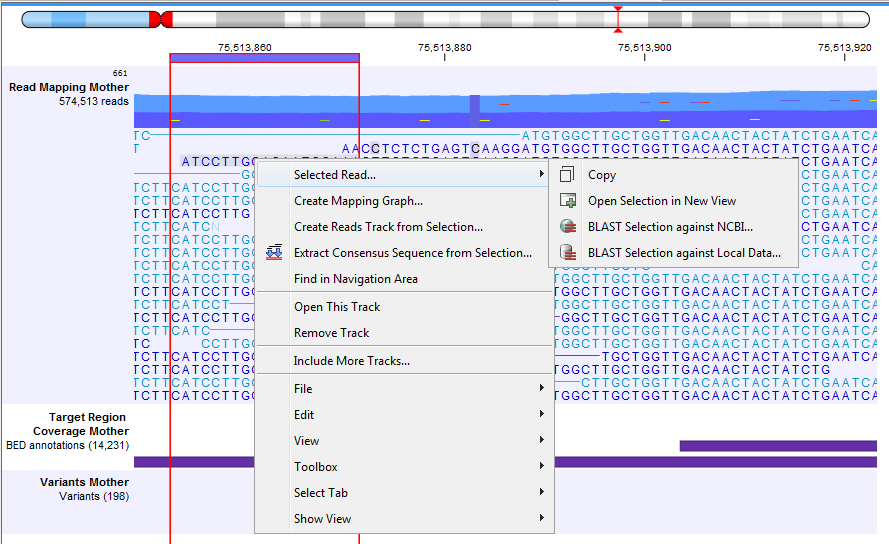
Figure 31.23: Adjusting the height of the track. - Hover the mouse cursor over a position in a reads track to reveal a tooltip with information about the reads supporting certain base calls, or a deletion, at that position, as well as the directions of those reads (figures 31.24 and 31.25). For overlapping paired reads that disagree, ambiguous bases are represented by their IUPAC codes. Use 'Show strands of paired reads' to show all bases from overlapping paired reads, see Overlapping paired reads for more information.
The tooltip uses the following symbols for the counts:
- + for single-end read mapped in forward direction, i.e., the number of green reads
- - for single-end read mapped in reverse direction, i.e., the number of red reads
- p+ for paired-end read mapped in forward direction (one count per pair), i.e., the number of dark blue reads
- p- for paired-end read mapped in reverse direction (one count per pair), i.e., the number of light blue reads
- ? for reads mapped in multiple places, i.e., the number of yellow reads
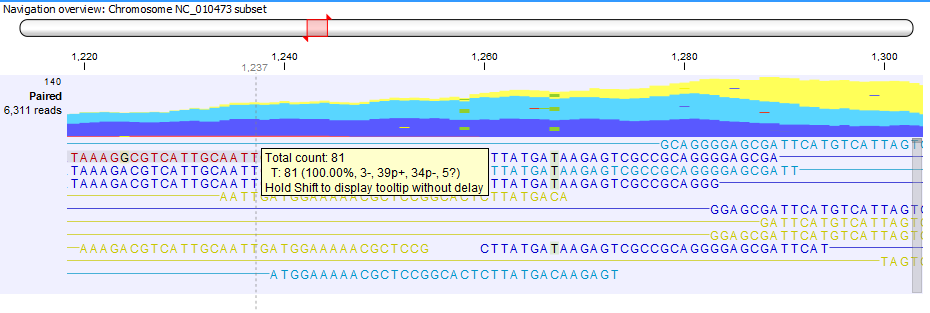
Figure 31.24: Example of tooltip information in a non-aggregated view of a reads track containing paired reads.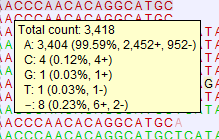
Figure 31.25: Example of tooltip information in a non-aggregated view of a reads track containing single reads. In this example, 8 reads support a deletion.Tip: With larger mappings, there can be a short delay before the tooltip appears. To speed this up, click on the Shift key while moving the mouse over the reads. Tooltips then appear without delay.
For information on the side panel settings for reads tracks, see Reads tracks Side Panel settings.
Subsections