Assemble sequences
Assemble Sequences takes as input reads, provided as sequences and sequence lists, assembling them into contigs without the need for a reference sequence. Assemble Sequences to Reference is recommended when a reference sequence is available.
The tool can assemble a maximum of 10,000 sequences at a time. To assemble a larger number of sequences, use De Novo Assembly.
To run the tool, go to:
Tools | Molecular Biology Tools (![]() ) | Sanger Sequencing Analysis (
) | Sanger Sequencing Analysis (![]() )| Assemble Sequences (
)| Assemble Sequences (![]() )
)
The following options can be configured (figure 23.6):
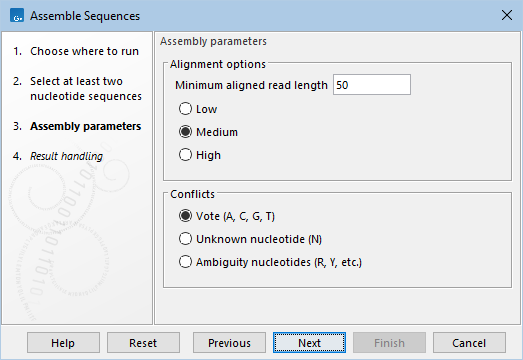
Figure 23.6: Options for Assemble Sequences.
- Minimum aligned read length The minimum number of nucleotides from a read sequence that must align successfully to the contig for it to be included in the assembly. Reads that do not meet this threshold are excluded.
- The alignment scoring stringency can be set to Low, Medium, or High. Higher values of stringency typically result in contigs with fewer ambiguities, but may exclude more reads and produce more, shorter contigs.
- Conflicts The following options are available for handling conflicts, i.e. positions where the aligned reads disagree on the nucleotide (A, C, T, or G):
- Vote (A, C, G, T) The most frequent nucleotide is used. If there is a tie, nucleotides are chosen in the order: A > C > G > T.
- Unknown nucleotide (N). The IUPAC ambiguity code 'N', representing any nucleotide, is used.
- Ambiguity nucleotides (R, Y, etc.). An appropriate IUPAC ambiguity code is used to reflect the nucleotide variation observed in the reads.
Read conflicts are annotated on the contigs.
The following output options can be configured:
- Create read mappings for contigs When checked, the read mappings are output, each consisting of an assembled contig and the aligned reads along with their trace data. See View and edit contigs and read mappings for more details.
- Create consensus sequences When checked, the assembled contig sequences are output as a sequence list, if multiple contigs are assembled, or a single sequence. Trace data is not retained.