Detecting DNA methylation
DNA-Methylation is one of the most significant epigenetic mechanisms for cell-programming. DNA-methylation alters the gene expression pattern such that cells can recall their cell type, essentially removing the necessity for continuous external signalling or stimulation. Even more, DNA-methylation is retained throughout the cell-cycle and thus inherited through cell division. DNA-Methylation involves the addition of a methyl group to the 5-position of the cytosine pyrimidine ring or the number 6 nitrogen of the adenine purine ring. DNA-methylation at the 5-position of cytosines typically occurs in a CpG dinucleotide context. CpG dinucleotides are often grouped in clusters called CpG islands, which are present in the 5' regulatory regions of many genes.
A large body of evidence has demonstrated that aberrant DNA-methylation is associated with unscheduled gene silencing, resulting in a broad range of human malignancies. Aberrant DNA-methylation manifests itself in two distinct forms: hypomethylation (a loss/reduction of methylation) and hypermethylation (an increase in methylation) compared to normal tissue. Hypermethylation has been found to play significant roles in carcinogenesis by repressing transcription of tumor suppressor genes, while hypomethylation is implicated in the development and the progression of cancer.
DNA methylation detection methods comprise various techniques. Early methods were based on restriction enzymes, cleaving either methylated or unmethylated CpG dinucleotides. These were laborious but suitable to interrogate the DNA-methylation status of individual DNA sites. It was later discovered that bisulfite treatment of DNA turns unmethylated cytosines into uracils while leaving methylated cytosines unaffected (figure 37.13). This discovery provided a more effective screening method that, in conjunction with whole genome shotgun sequencing of bisulfite converted DNA, opened up a broad field of genome-wide DNA methylation studies. Ever since, bisulfite shotgun sequencing (BS-seq) is considered a gold-standard technology for genome-wide methylation profiling at base-pair resolution.
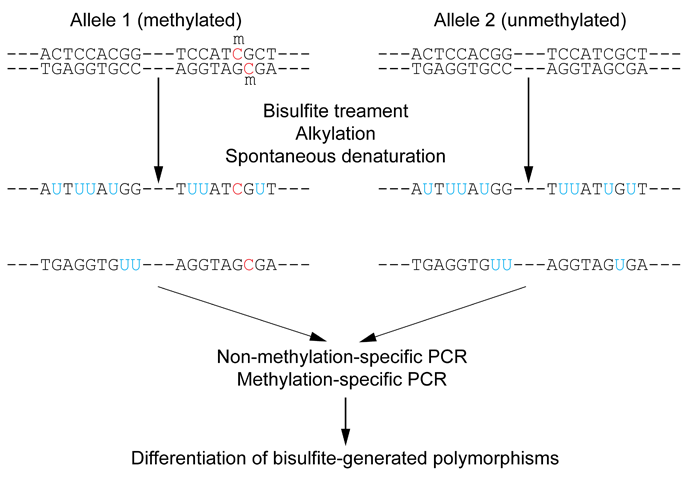
Figure 37.13: Outline of bisulfite conversion of sample sequence of genomic DNA. Nucleotides in blue are unmethylated cytosines converted to uracils by bisulfite, while red nucleotides are 5-methylcytosines resistant to conversion. Source: https://en.wikipedia.org/wiki/Bisulfite_sequencing
Figure 37.14 depicts the conceptual steps of the Bisulfite-sequencing:
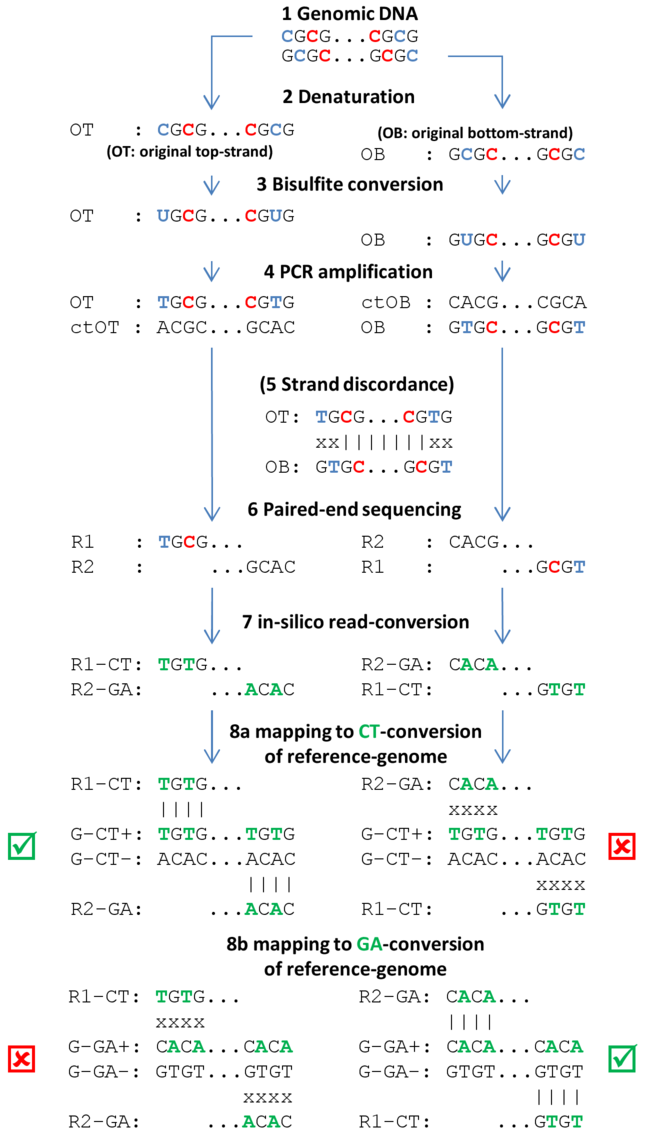
Figure 37.14: Individual steps of BS-seq workflow include denaturation of fragmented sample DNA, bisulfite conversion, subsequent amplification, sequencing and mapping of resulting DNA-fragments. (See text for explanations). Methylated cytosines are drawn in red, unmethylated cytosines and respective uracils/thymidines in blue. DNA-nucleotides that are in-silico converted (during read mapping) are given in green.
- Genomic DNA Genomic DNA is extracted from cells, sheared to fragments, end-repaired,
size-selected (around 400 base pairs depending on targeted read length) and
ligated with Illumina methylated sequencing adapters. End-repair involves
either methylated or unmethylated cytosines, possibly skewing true methylation
levels. Therefore, 3'- and 5'-ends of sequenced fragments should be
soft-clipped prior to assessing methylation levels.
- Denaturation Fragments must be denatured (and kept denatured during bisulfite conversion), because bisulfite can only convert single-stranded DNA.
- Bisulfite conversion Bisulfite converts unmethylated cytosines into uracils, but leaves methylated cytosines unchanged. Because bisulfite conversion has degrading effects on the sample DNA, the conversion duration is kept as short as possible, sometimes resulting in non-complete conversions (i.e. not all unmethylated cytosines are converted).
- PCR amplification PCR-amplification reconstructs the complementary strands of the converted
single-stranded fragments and turns uracils into thymines.
- Strand discordance Not an actual step of the workflow, but to illustrate that bisulfite
converted single-stranded fragments are not reverse-complementary anymore after
conversion.
- Paired-end sequencing Directional paired-end sequencing yields read pairs from both strands of
the original sample-DNA. The first read of a pair is known to be sequenced
either from the original-top (OT) or the original-bottom (OB) strand. The second
read of a pair is sequenced from a complementary strand, either ctOT
or ctOB. It is a common misunderstanding that the first read of a pair yields
methylation information for the top-strand and the second read for the
bottom-strand (or vice versa). Rather, both reads of a read pair report
methylation for the same strand of sample DNA, either the top or the bottom
strand. Individual read pairs can of course arise from both the top and the
bottom strand, eventually yielding information for both strands of the sample
DNA.
- In silico read-conversion The only bias-free mapping approach for BS-seq reads involves in-silico
conversion of all reads. All cytosines within all first reads of a pair are
converted to thymines and all guanines in all second reads of a pair are
converted to adenines (complementary to C-T conversion).
- Mapping to CT- or GA-conversion of reference genome The reference genome is also converted into two different in silico versions. In the first conversion all cytosines are replaced by thymines and, in the second conversion, all guanines are converted to adenines. The in silico converted read pairs are then independently mapped to the two conversions of the reference genome and the better of the two mappings is reported as final mapping result (see green checkboxes).
Note: with non-directional BS-seq, no assumptions regarding the strand origins of either of the reads of a pair can be made (see step 6). Therefore, two different conversions of the read pair need to be created: the first read of a converted pair consists of the CT-conversion of read 1 and the GA-conversion of read 2, and the second converted pair consists of the GA-conversion of read 1 and the CT-conversion of read 2. Both converted reads pairs are subsequently mapped to the two conversion of the reference genome. The best out of the four resulting mappings is then reported as the final mapping result.