Import tracks
Tracks (see Tracks) are imported in a special way, because extra information is needed in order to interpret the files correctly.
To import tracks, click on the Import (![]() ) icon in the Toolbar and choose Tracks. This will open a dialog as shown in figure 7.2.
) icon in the Toolbar and choose Tracks. This will open a dialog as shown in figure 7.2.
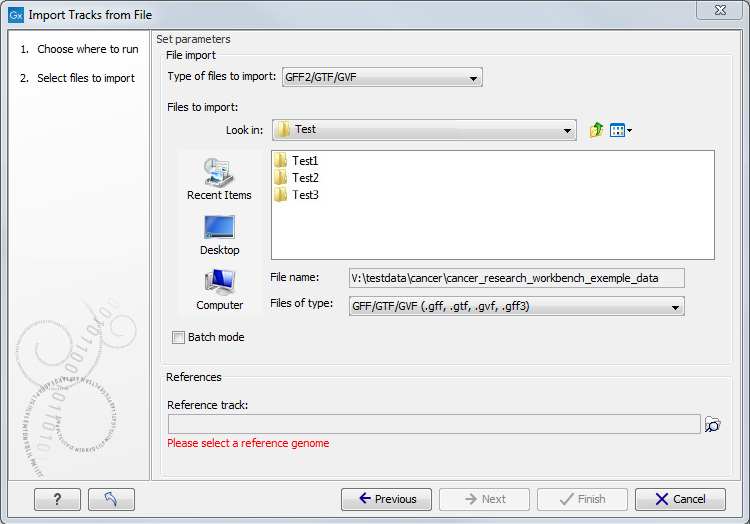
Figure 7.2: Select files to import.
At the top, you select the file type to import. Below, select the files to import. If import is performed with the batch option selected, then each file is processed independently and separate tracks are produced for each file. If the batch option is not selected, then variants for all files will be added to the same track (or tracks in the case VCF files including genotype information). The formats currently accepted are:
- FASTA
- This is the standard fasta importer that
will produce a sequence track rather than a standard fasta sequence. Please
note that this could also be achieved by importing using Standard Import (see Standard import).
and subsequently converting the sequence or sequence list to a track (see Converting data to tracks and back).
- GFF2/GTF/GVF
- A GFF2/GTF
file does not contain any sequence information, it only contains a list of
various types of annotations. A GVF file is similar to a GFF file but uses
Sequence Ontology to describe genome variation data (see https://github.com/The-Sequence-Ontology/Specifications/blob/master/gvf.md). For these formats, the importer
adds the annotation in each of the lines in the file to the chosen sequence,
at the position or region in which the file specifies that it should go, and
with the annotation type, name, description etc. as given in the file.
However, special treatment is given to annotations of the types CDS, exon,
mRNA, transcript and gene.
For these, the following applies:
- A gene annotation is generated for each gene_id. The region annotated extends from the leftmost to the rightmost positions of all annotations that have the gene_id (gtf-style).
- CDS annotations that have the same transcriptID are joined to one CDS annotation (gtf-style). Similarly, CDS annotations that have the same parent are joined to one CDS annotation (gff-style).
- If there is more than one exon annotation with the same transcriptID these are joined to one mRNA annotation. If there is only one exon annotation with a particular transcriptID, and no CDS with this transcriptID, a transcript annotation is added instead of the exon annotation (gtf-style).
- Exon annotations that have the same parent mRNA are joined to one mRNA annotation. Similarly, exon annotations that have the same parent transcript, are joined to one transcript annotation (gff-style).
For a comprehensive source of genomic annotation of genes and transcripts, we refer to the Ensembl web site at https://www.ensembl.org/info/data/ftp/index.html. On this page, you can download GTF files that can be used to annotate genomes for use in other analyses in the CLC Genomics Workbench. You can also read more about these formats at https://gmod.org/wiki/GFF3, https://www.ensembl.org/info/website/upload/gff.html, and https://genomebiology.biomedcentral.com/articles/10.1186/gb-2010-11-8-r88.
- GFF3
- A GFF3 file contains a list of various types of annotations that
can be linked together with "Parent" and "ID" tags. Learn more about how the
CLC Genomics Workbench handles GFF3 format in GFF3 format.
- VCF
- This is the file format used for variants by the 1000 Genomes
Project and it has become a standard format. Read about VCF format here https://samtools.github.io/hts-specs/VCFv4.2.pdf. Learn how to access data at
https://www.internationalgenome.org/data/#DataAccess. Learn more about how the
CLC Genomics Workbench handles VCF format in VCF format.
- BED
- This format is typically used for simple annotations, such as target regions for sequence capture methods.
The format is described at https://genome.ucsc.edu/FAQ/FAQformat.html#format1.
The 3 required BED fields (chrom, chromStart and chromEnd) must be present as the first 3 columns in the file to be imported.
Optional BED fields, present in the order stipulated in the UCSC format, are also imported, with the exceptions listed below.
If there are additional columns, these are imported and assigned the header "Var" followed by a number, e.g. Var1, Var2, etc.
Exceptions:
- The itemRgb field is ignored.
- The thickStart and thickEnd fields are used to check if the annotation is valid, but are otherwise ignored.
- Wiggle
- The Wiggle format as defined by UCSC
(https://genome.ucsc.edu/goldenPath/help/wiggle.html) is used to
hold continuous data like conservation scores, GC content etc. When imported
into the CLC Genomics Workbench, a graph track is created. An example of a popular
Wiggle file is the conservation scores from UCSC which can be download for
human from
https://hgdownload.cse.ucsc.edu/goldenPath/hg19/phastCons46way/.
- UCSC variant database table dump
- Table dumps of variant
annotations from the UCSC can be imported using this option. Mainly files
ending with
.txt.gzon this list can be used: https://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/. Please note that importer is for variant data and is not a general importer for all annotation types. This is mainly intended to allow you to import the popular Common SNPs variant set from UCSC. The file can be downloaded from the UCSC web site here: https://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/snp138Common.txt.gz. Other sets of variant annotation can also be downloaded in this format using the UCSC Table Browser. - COSMIC variation database
- This lets you import the COSMIC database, which is a well-known publicly available primary
database on somatic mutations in human cancer. The file can be downloaded from the COSMIC web site here:
https://cancer.sanger.ac.uk/cancergenome/projects/cosmic/download,
You must first register to download the database. The following tsv format COSMIC files can be imported using the option COSMIC variation database under Import->Tracks:
- COSMIC Targeted Screens Mutants:
- Cosmic_CompleteTargetedScreensMutant_vXXX_GRCh37.tsv
- Cosmic_CompleteTargetedScreensMutant_vXXX_GRCh38.tsv
- COSMIC Genome Screen Mutants:
- Cosmic_GenomeScreensMutant_vXXX_GRCh37.tsv
- Cosmic_GenomeScreensMutant_vXXX_GRCh38.tsv
- COSMIC Census Genes Mutations
- Cosmic_MutantCensus_vXXX_GRCh37.tsv
- Cosmic_MutantCensus_vXXX_GRCh38.tsv
- COSMIC Resistance Mutations:
- Cosmic_ResistanceMutations_vXXX_GRCh37.tsv
- Cosmic_ResistanceMutations_vXXX_GRCh38.tsv
Variants in recent COSMIC tsv format files are 3'-shifted relative to the plus-strand of the reference. To compare variants detected using the CLC Genomics Workbench with COSMIC variants, it may be preferable to import COSMIC VCF files with variants 5'-shifted using the VCF importer. This is because variants detected using the CLC Genomics Workbench, in accordance with VCF recommendations. (See Gap placement.)
- COSMIC Targeted Screens Mutants:
Please see Annotation and variant formats for more information on how different formats (e.g. VCF and GVF) are interpreted during import in CLC format. For all of the above, zip files are also supported. Please note that for human data, there is a difference between the UCSC genome build and Ensembl/NCBI for the mitochondrial genome. This means that for the mitochondrial genome, data from UCSC should not be mixed with data from other sources (see https://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/). Most of the data above is annotation data and if the file includes information about allele variants (like VCF, Complete Genomics and GVF), it will be combined into one variant track that can be used for finding known variants in your experimental data.
For all types of files except fasta, you need to select a reference track as well. This is because most the annotation files do not contain enough information about chromosome names and lengths which are necessary to create the appropriate data structures.
Subsections