Remove Marginal Variants
Variant calling is always a balance between sensitivity and specificity. Remove Marginal Variants is designed for the removal of potential false positive variants. The tool can be configured to remove variant calls with low frequency, a skewed forward-reverse reads balance, or those predominantly supported by low quality bases.
A new variant track is produced as output, leaving the original variant track as it was.
To run Remove Marginal Variants, go to:
Tools | Resequencing Analysis (![]() ) | Variant Filtering (
) | Variant Filtering (![]() ) | Remove Marginal Variants (
) | Remove Marginal Variants (![]() )
)
This opens a dialog where you can select a variant track (![]() ).
).
Click on Next to move to the dialog where filtering thresholds can be set, as shown in figure 32.2.
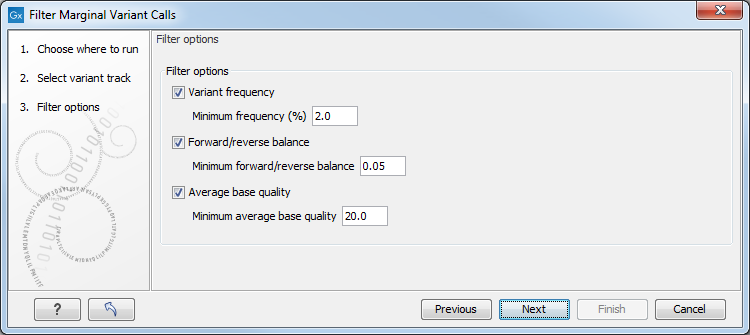
Figure 32.2: One or more thresholds can be configured, defining the basis for variant removal.
The following thresholds can be specified. All alleles are investigated separately.
- Variant frequency. The frequency filter will remove all variants having alleles with a frequency (= number of reads supporting the allele/number of all reads) lower than the given threshold.
- Forward/reverse balance. The forward/reverse balance filter will remove all variants having alleles with a forward/reverse balance of less than the given threshold.
- Average base quality. The average base quality filter will remove all variants having alleles with an average base quality of less than the given threshold.
If several thresholds are applied, just one needs to be fulfilled to discard the allele. For more information about how these values are calculated, please refer to Variant tracks.
If all non-reference alleles at a position are removed, any remaining homozygous reference alleles will also be removed at that position.
A new variant track is produced by this tool, containing just the variants that exceeded the configured thresholds.