Sanger sequencing data
The Sanger importer is designed to import large volumes of Sanger high-throughput sequencing data. Formats supported are ab, abi, ab1, scf and phd. Uncompressed files as well as files compressed using gzip (.gz), zip (.zip) or bzip2 (.bz2) can be provided as input.
To launch the Sanger importer, go to:
Import (![]() ) | Sanger (
) | Sanger (![]() ).
).
This opens a dialog where files can be selected and import options specified (figure 7.22).
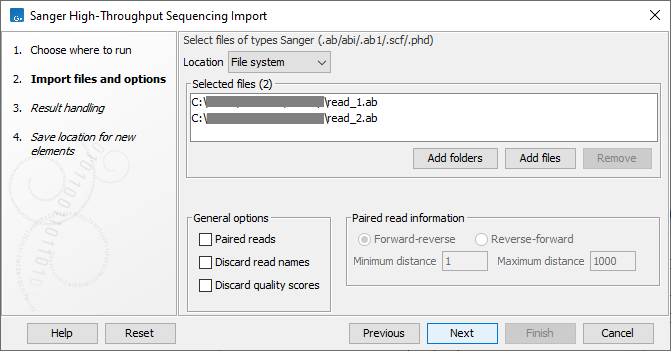
Figure 7.22: Importing Sanger sequencing data.
Sanger sequencing data can also be imported using the Standard Import (![]() ) (see Standard import). The following are key differences of the high-throughput importer when compared to the Standard Import:
) (see Standard import). The following are key differences of the high-throughput importer when compared to the Standard Import:
- It is designed to handle large volumes of data efficiently.
- A given batch of sequences is imported to a single sequence list. The Standard Import creates a single sequence element for each imported sequence.
- The chromatogram traces are removed (quality scores remain). This improves performance; trace data takes up a lot of disk space, and this can impact speed and memory consumption of downstream analyses.
- Paired reads are supported.
Sanger data can also be imported using the on-the-fly import functionality available in workflows, described in Launching workflows individually and in batches. Both the Sanger importer and the Standard Import ("Trace files") are available using the on-the-fly import.
The General options to the left are:
- Paired reads Import pairs of reads into a single sequence list. When enabled, the files selected for import are sorted, and then the first and second file are imported together as paired reads, the third and fourth file are imported together as paired reads, etc. The selection of "Forward-reverse" or "Reverse-forward" in the "Paired read information" area determines whether the first file is treated as containing forward reads and the second file reverse reads, or vice versa. As an example, with two files:
sample1_fwdcontaining forward reads andsample1_revcontaining reverse reads, and selecting the "Forward-reverse" option, you would get a single sequence list, marked as containing paired reads, with the pairs in the expected orientation. Insert sizes can also be specified, using the "Minimum distance" and "Maximum distance" settings. Data sets with different insert sizes should be imported separately. Read more about handling paired data. - Discard read names Selecting this option saves disk space. Names of individual sequences are often irrelevant in large datasets.
- Discard quality scores Selecting this option can save substantial space, and can decrease memory consumption for downstream activities. Quality scores should be retained if they are relevant to your work. For example, quality scores are used for variant detection and can (optionally) be seen displayed in views of read mappings.
The next wizard step provides some options for handling the results. When the option to "Create subfolders per batch unit" is enabled, each sequence list created is put into its own subfolder. This can be helpful for running analyses in batches and for organizing the results of subsequent analyses.