Import with Metadata
Using the Import with Metadata template workflow, sequence data is imported, and a CLC Metadata Table is created containing the metadata provided in an Excel, CSV or TSV format file. Each imported sequence list is associated with the row in that CLC Metadata Table that contains metadata relating to it.
This is a very simple workflow, containing just an Iterate element connected to an Input element and an Output element (figure 15.94).
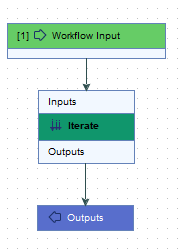
Figure 15.94: The Import with Metadata template workflow
The following features of workflows are key to how this works:
- A CLC Metadata Table with a row recording each workflow output can be created. This element is named "Workflow Result Metadata". Using this template workflow, it will contain one row per imported sequence list.
The Workflow Result Metadata element is described in more detail in the section about launching workflows.
- When batch units are defined using metadata, all the columns in the metadata file are included in the Workflow Result Metadata element.
Together, these features allow this simple workflow to import data, create a CLC Metadata Table containing information about the data being imported, and establish associations between the imported data and the CLC Metadata Table.
This template workflow is configured to import only sequence data. However, import of other sorts of data can easily be configured by copying the template workflow and editing it.
Launching the workflow
The Import with Metadata template workflow is at:
Workflows | Template Workflows | Preparing Raw Data | Import with Metadata (![]() )
)
In the first step, select the format of the data being imported and check the options are configured as needed.
Batch units should be defined using metadata stored in an Excel, CSV or TSV format file. Requirements for this file are described in Defining batch units based on metadata.
Once the metadata file has been selected, the column to use to define the batch units can be specified.
In the Batch overview wizard step, the organization of the input files into batch units can be reviewed (figure 15.95).
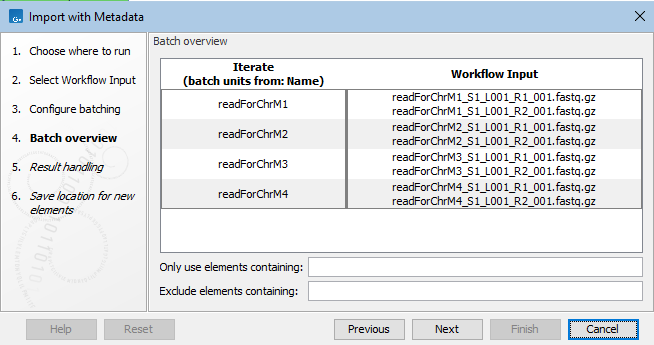
Figure 15.95: In the batch overview step, you can check that input data has been grouped into batch units as expected.
In the Result handling step, ensure the option Create workflow result metadata is checked so the "Workflow Result Metadata" element will be created.
An example of the results of this workflow is shown in figure 15.96).
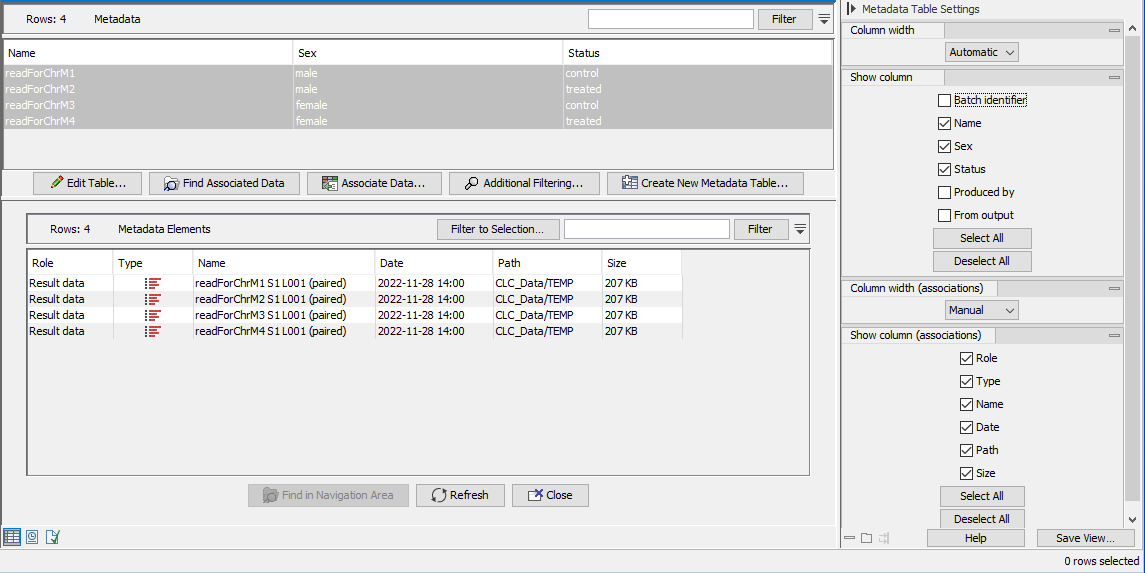
Figure 15.96: The CLC Metadata Table created using the Import with Metadata template workflow has been opened. There is a row per sequence list imported. In this view, some column names in the side panel have been unchecked so that only the sample-specific information is shown. The sequence lists associated with the metadata rows are listed in the bottom panel as a result of selecting all the rows and clicking on the Find Associated Data button.