Map Long Reads to Reference
The Map Long Reads to Reference tool enables aligning long reads to a reference with minimap2 [Li, 2018].
To run the tool, go to:
Tools | Resequencing Analysis (![]() ) | Map Long Reads to Reference (
) | Map Long Reads to Reference (![]() )
)
Select one or more sequence lists containing long reads.
In the References dialog, select one or more reference sequences. You can select either individual sequences, a list of sequences or a sequence track as reference (figure 31.12).
Figure 31.12: Specifying the reference sequences and masking.
The next part of the dialog lets you mask the reference. Masking means that selected regions of the reference are ignored during read mapping. Reads will not be mapped to these regions, but the full reference is still included in the output.
Masking can be useful when reads are expected to originate only from specific regions, for example when working with targeted sequencing data. However, masking should be used with care. If reads originate outside the selected regions, they may be mapped to less suitable locations, which can affect downstream analyses such as variant detection.
Masking large numbers of regions, such as repetitive sequences, is generally not recommended. Repeats are handled automatically during mapping, and masking them may reduce performance and lead to incorrect read placement.
To mask a reference using regions defined in a masking track, choose:
- Include annotated only to map reads only to those regions.
- Exclude annotated to ignore those regions.
If your regions are stored as sequence annotations, they can be converted to a track.
In the Mapping options dialog, set the read mapping parameters (figure 31.13):
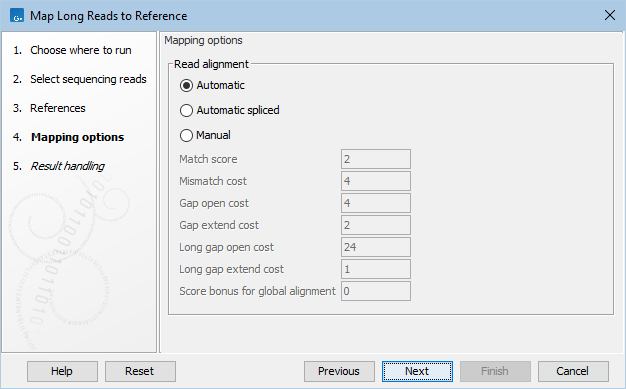
Figure 31.13: Map Long Reads to Reference mapping options.
- Mapping mode. Choose between the following modes for parameter setting:
- Automatic. Match cost parameters are set automatically based on the read type of the first input (e.g. Oxford Nanopore or Pacbio HiFi).
- Automatic spliced. Similar to Automatic, except it generates spliced alignments, which can be useful for visualizing RNA-seq data.
- Manual. Allows match costs to be specified manually. This will overwrite the read type specific match costs that would otherwise be used with the values entered below.
- Match score. Score of a match.
- Mismatch cost. Cost of a mismatch.
- Gap open cost. Cost of starting a gap.
- Gap extend cost. Cost of extending a gap.
- Long gap open cost. Cost of starting a long gap. Long gaps are typically more expensive to open but cheaper to extend. The alignment will use the cheaper alternative.
- Long gap extend cost. Cost of extending a long gap.
- Score bonus for global alignment. A bonus that may be added to the alignment score if the alignment encompasses all nucleotides of the read.
For guidance on adjusting match cost parameters, see Mapping parameters. Additional information on parameters are available from the minimap2 documentation.
Subsections