Extract Consensus Sequence
Extract Consensus Sequence takes as input stand-alone read mappings, read tracks, alignments, or nucleotide BLAST results, and outputs a consensus sequence.
Consensus sequences can also be extracted directly from elements opened in the View Area by selecting Extract New Consensus Sequence (![]() ) from the right-click menu on:
) from the right-click menu on:
- For stand-alone read mappings: the reference sequence or a selection within it, or the consensus sequence.
- For alignments: the consensus sequence.
- For nucleotide BLAST results: a selection within the query sequence.
To run the tool, go to:
Tools | Resequencing Analysis (![]() ) | Extract Consensus Sequence (
) | Extract Consensus Sequence (![]() )
)
The following options for extracting the consensus sequence can be configured (figure 31.47):
- Threshold Coverage above this value is considered high, while coverage at or below it is considered low. Overlapping paired-end reads count as two when calculating the coverage.
A higher threshold yields a more reliable consensus but reduces completeness.
- Low coverage handling Options for handling regions with low coverage.
- Remove regions with low coverage No consensus is generated in regions of low coverage.
- Join after removal High coverage regions are joined into a single consensus sequence.
- Split into separate sequences Each high coverage region produces an individual consensus sequence.
- Insert 'N' ambiguity symbols The consensus is set to 'N' at each position with low coverage.
- Fill from reference sequence The consensus is set to the reference symbol at each position with low coverage.
- Remove regions with low coverage No consensus is generated in regions of low coverage.
- Conflict resolution Options for resolving read disagreements at individual positions within high coverage regions.
- Vote The consensus is set to the most supported symbol (see Use quality score), excluding ambiguous symbols.
In case of a tie, symbols are chosen in the order: A > C > G > T.
To preserve biological heterozygous variation, see Insert ambiguity codes.
- Insert ambiguity codes The consensus is set to the IUPAC ambiguity code that best reflects the variation observed in the reads.
The following options determine which symbols contribute to the ambiguity codes:
- Noise threshold Only symbols with support greater than this value (see Use quality score) contribute to the ambiguity code.
- Minimum nucleotide count Only symbols present in at least this number of reads contribute to the ambiguity code.
Positions where no symbol qualifies to contribute to the ambiguity code are not included in the consensus.
- Noise threshold Only symbols with support greater than this value (see Use quality score) contribute to the ambiguity code.
- Vote The consensus is set to the most supported symbol (see Use quality score), excluding ambiguous symbols.
- Use quality score The support of a symbol is determined by:
- When unchecked: the percentage of reads containing the symbol.
- When checked: the sum of the symbol's quality scores, expressed as a percentage of the total quality score at that position.
- Consensus sequence decoration Options for annotating the consensus sequence.
- Add consensus annotations (conflicts, indels, low coverage etc.) When checked, annotations are added to the consensus sequence to indicate resolved conflicts, deletions relative to the reference, and low coverage regions, provided the Split into separate sequences option is not selected.
For inputs containing many reads or long references, many such annotations may be generated.
- Keep annotations already on consensus and Transfer annotations from reference When checked, annotations present on the consensus or on the reference in the input stand-alone read mapping are copied to the extracted consensus sequence. The copied annotations are placed in regions corresponding to their original location in the input data, although actual coordinates may differ. Annotations may be split if the Split into separate sequences option is selected.
These options are not enabled for types of input other than stand-alone read mapping.
- Add consensus annotations (conflicts, indels, low coverage etc.) When checked, annotations are added to the consensus sequence to indicate resolved conflicts, deletions relative to the reference, and low coverage regions, provided the Split into separate sequences option is not selected.
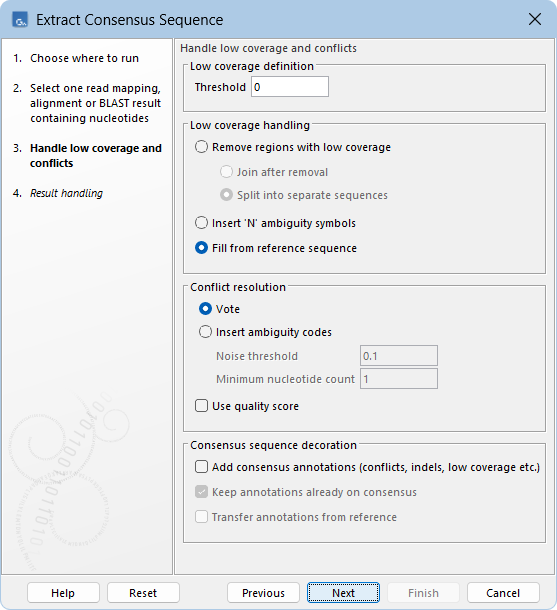
Figure 31.47: Options for extracting a consensus sequence.
Quality scores on the consensus sequence
When quality scores are present in the input, they are propagated to the extracted consensus as follows.
Consider a consensus symbol ![]() , and let
, and let ![]() and
and ![]() be quality scores sums at its position, defined as:
be quality scores sums at its position, defined as:
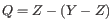 : The sum of quality scores from all reads.
: The sum of quality scores from all reads.
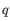 : The sum of quality scores from reads supporting
: The sum of quality scores from reads supporting 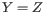 , defined as:
, defined as:
- Reads containing the symbol , if the Vote option is selected.
- Reads containing symbols that contribute to the ambiguity code, if the Insert ambiguity codes option is selected.
- Reads containing the symbol
The consensus symbol ![]() is assigned a quality score
is assigned a quality score
![]() , bounded to a minimum of 0 and a maximum of 64.
, bounded to a minimum of 0 and a maximum of 64.