General notes on handling paired data
During import, information about paired data (orientation and distances) can be specified. This information is stored and can be taken into account during analyses.
The members of each read pair are placed sequentially in a sequence list. E.g. for paired-end data, the forward read in a pair is followed by the reverse read in that pair. When manipulating sequence lists, it is important not to interfere with this ordering. If read names were not discarded during import, the members of each read pair should have the same name, except for a 1 or a 2 somewhere in the name.
This information is used by tools such as Map Reads to Reference and RNA-Seq Analysis. Some tools have an "Auto-detect paired distances" option. When this option is checked, the distances set during import are disregarded, and are instead calculated from the data (see Mapping paired reads).
The orientation and distance values for a sequence list can be edited by opening the Element Info (![]() ) view of the sequence list (see Element information view). If the "Paired sequences" box is unchecked, the sequences will be handled as single (non-paired) reads.
) view of the sequence list (see Element information view). If the "Paired sequences" box is unchecked, the sequences will be handled as single (non-paired) reads.
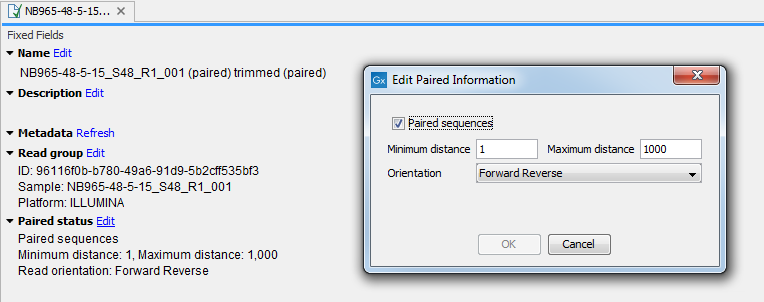
Figure 7.18: Editing paired orientation and distance in the Element Info view.
A paired read distance includes the full read sequence. E.g. for paired-end reads, the full read sequence is from the beginning of the forward read to the beginning of the reverse read (figure 7.19).
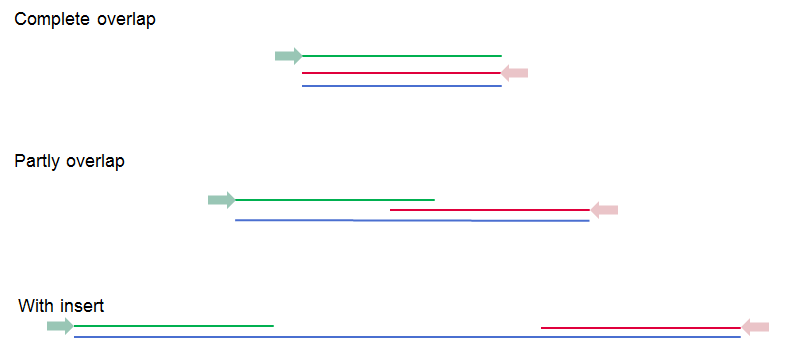
Figure 7.19: Green lines represent forward reads, red lines reverse reads, and in blue is shown the distance of the sequenced DNA fragment. Thus, if there is a complete overlap, the minimum distance will not be 0, but the length of the overlap.