Guided realignment
One limitation of the local realignment algorithm employed is that at least one read must be aligned correctly according to the true indel present in the data. If none of the reads is aligned correctly, local realignment cannot improve the alignment, since it lacks information about how to do so. To overcome this limitation, local realignment can be guided in two ways:
- Guidance variants: By supplying the Local realignment tool with a track of guidance variants. There are two modes for using the guidance variant track: either the 'un-forced' guidance mode (if the 'Force realignment to guidance-variants' is left un-ticked) or the 'forced' guidance mode (if the 'Force realignment to guidance-variants' is ticked).
In the 'unforced' mode, 'pseudo-reads' are given to the local realignment algorithm representing the guidance variants, allowing the local realignment algorithm to explore the paths in the graph corresponding to these alignments.
A scoring scheme where alignment to reference is preferred, is employed during first realignment pass, to determine the initial read support for the guidance variants. When more than one realignment pass is selected, the additional realignment passes are carried out using the standard scoring scheme where the most frequently used alignment path is preferred, and a supplementary limited realignment pass is performed in regions with guidance variants, to make up for the different scoring scheme used during first realignment pass.
In the 'forced' mode, 'pseudo-references' are given to the local realignment algorithm representing the guidance variants, allowing the reads to be aligned to allele sequences of these in addition to the original reference sequence - with matches being awarded and encouraged equally much. The 'unforced' mode can be used with any guidance variant track as input. The 'force' mode should only be used with guidance variants for which there is strong prior evidence that they exist in the data (e.g., the 'InDel' track from the Structural Variants' tool (see Section 32.10) produced on the read mapping that is being aligned). Unless you do have strong evidence for the presence of these guidance variants, we do not recommend using the 'forced' mode as it can lead to the introduction of false positives in your alignment and all subsequent analyses.
- Concurrent local realignment of multiple samples: Multiple input read mappings increase the chance to encounter at least one read mapped correctly. This guiding mechanism has been particularly designed for scenarios, where samples are known to be related, such as in family trials.
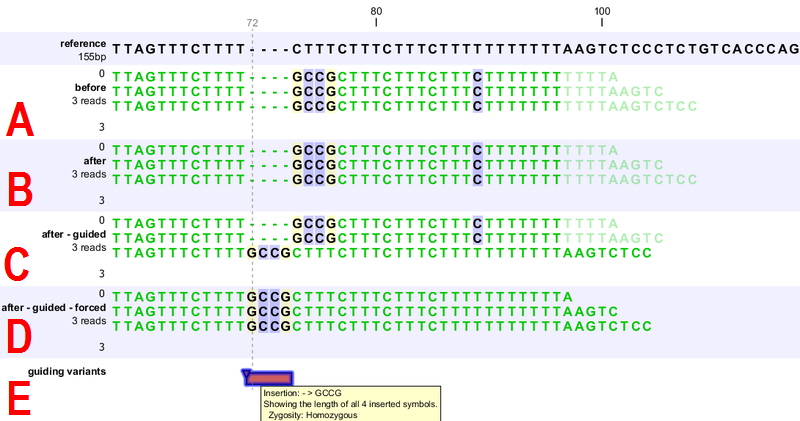
Figure 31.40: [A] Three reads are misaligned in the presence of a four nucleotide insertion relative to the reference. [B] When applying local realignment without guidance the alignment is not improved. [C] Here local realignment is performed in the presence of the guiding variant track seen in (E). This enables the algorithm to consider alternative alignments, which are accepted whenever they have significant improvements over the original (as in read three that has a comparatively long unaligned-end). [D] If the alignment is performed with the option "Force realignment to guidance-variants" enabled, the realignment will be forced to realign according to the guiding variant track shown in (E), and this will result in realignment of all three reads. [E] The guiding variant track contains, amongst others, the four nucleotide insertion.

Figure 31.41: [B] Three reads are misaligned in the presence of a four nucleotide insertion into the reference. Applying local realignment without guiding information would not yield any improvements (not shown). [C] Performing local realignment on both samples (A) and (B) enables the algorithm to improve the alignments of sample (B).