Oxford Nanopore
The Oxford Nanopore importer supports import of reads generated by Oxford Nanopore sequencing technology.
To launch the Oxford Nanopore importer, go to:
Import (![]() ) | Oxford Nanopore (
) | Oxford Nanopore (![]() ).
).
This opens a dialog where files can be selected and import options specified (figure 7.10).
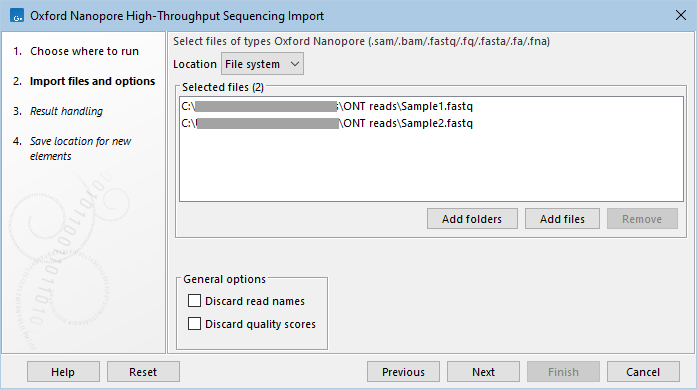
Figure 7.10: Importing data from Oxford Nanopore systems.
The following file formats are supported:
- Fastq (
.fastq/.fq). Uncompressed files as well as files compressed using gzip (.gz), zip (.zip) or bzip2 (.bz2) can be provided as input. Quality scores are expected to be in the NCBI/Sanger format, see Quality scores in the Illumina platform. The importer processes UMI information from the fastq read headers, see General notes on UMIs. - Fasta files (.fasta) which contain sequence data. Compressed Fasta (.fasta.gz) files are also supported.
- SAM or BAM files (.sam/.bam). Mapping information in the file is disregarded.
The General options are:
- Discard read names. Read names can be discarded to save disk space without affecting analysis results. Keeping read names can be useful in some circumstances, such as when inspecting sequence list contents or when working downstream with subsets of sequences.
- Discard quality scores. Quality scores are visible in read mappings and are used by various tools, e.g. for variant detection. If quality scores are not relevant, use this option to discard them and reduce disk space and memory consumption.