Run the InDels and Structural Variants tool
Go to:
Tools | Resequencing Analysis (![]() ) | Variant Detection (
) | Variant Detection (![]() ) | InDels and Structural Variants (
) | InDels and Structural Variants (![]() )
)
This will open up a dialog. Select the read mapping of interest as shown in figure 32.38 and click on the button labeled Next.
Figure 32.38: Select the read mapping of interest.
The next wizard step (Figure 32.39) is concerned with specifying parameters related to the algorithm used for calling structural variants. The algorithm first identifies positions in the mapping(s) with an excess of reads with left (or right) unaligned ends. Once these positions and the consensus sequences of the unaligned ends are determined, the algorithm maps the determined consensus sequences to the reference sequence around other positions with unaligned ends. If mappings are found that are in accordance with a 'signature' of a structural variant, a structural variant is called.
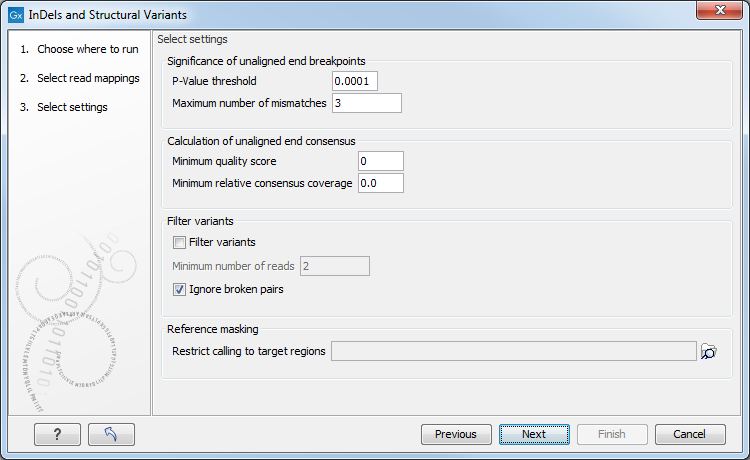
Figure 32.39: Select the relevant settings.
The 'Significance of unaligned end breakpoints' parameters are concerned with when a position with unaligned ends should be considered by the algorithm, and when it should be ignored:
- P-value threshold: Only positions in which the fraction of reads with unaligned ends is sufficiently high will be considered. The 'P-value threshold' determines the cut-off value in a Binomial Distribution for this fraction. The higher the P-value threshold is set, the more unaligned breakpoints will be identified.
- Maximum number of mismatches: The 'Maximum number of mismatches' parameter determines which reads should be considered when inferring unaligned end breakpoints. Poorly map reads tend to have many mis-matches and unaligned ends, and it may be preferable to let the algorithm ignore reads with too many mis-matches in order to avoid false positives and reduce computational time. On the other hand, if the allowed number of mis-matches is set too low, unaligned end breakpoints in proximities of other variants (e.g. SNVs) may be lost. Again, the higher the number of mis-matches allowed, the more unaligned breakpoints will be identified.
The Calculation of unaligned end consensus parameters can improve the calculation of the unaligned end consensus by removing bases according to:
- Minimum quality score: quality score under which bases should be ignored.
- Minimum relative consensus coverage: consensus coverage threshold under which bases should be ignored. The relative consensus coverage is calculated by taking the coverage at the current nucleotide position and dividing by the maximum coverage obtained along the unaligned ends upstream from this position. When the value thus calculated falls below the specified threshold, consensus generation stops. The idea behind the "Minimum relative consensus coverage" option is to stop consensus generation when dramatic drops in coverage are observed. For example, a drop from 1000 coverage to 10 coverage would give a relative consensus coverage of 10/1000 = 0.01.
The 'Filter variants' parameters are concerned with the amount of evidence for each structural variant required for it to be called:
- Filter variants: When the Filter variants box is checked, only variants that are inferred by breakpoints that together are supported by at least the specified Minimum number of reads will be called.
- Ignore broken pairs: This option is checked by default, but it can be unchecked to include variants located in broken pairs.
'Reference masking' allows specification of target regions:
- Restrict calling to target regions: When specifying a target region track only reads that overlap with at least one of the targets will be examined when the unaligned end breakpoints are identified. Hence only breakpoints that fall within, or in close proximity of, the targets will be identified (a read may overlap a target, but have an unaligned end outside the target - these are also identified and therefore breakpoints outside, but in the proximity of the target). The runtime will be decreased when you specify a target track as compared to when you do not.
Note! As the set of identified unaligned end breakpoints differs between runs where a target region track has been specified and where it has not, the set of predicted indels and structural variants is also likely to differ. This is because the indels and structural variants are predicted from the mapping patterns of the unaligned ends at the set of identified breakpoints. This is also the case even if you restrict the comparison to only involve the indels and structural variants detected within the target regions. You cannot expect these to be exactly the same but you can expect a large overlap.
Specify these settings and click Next. The "Results handling" dialog (Figure 32.40) will be opened. The Indels and Structural Variants tool has the following output options:
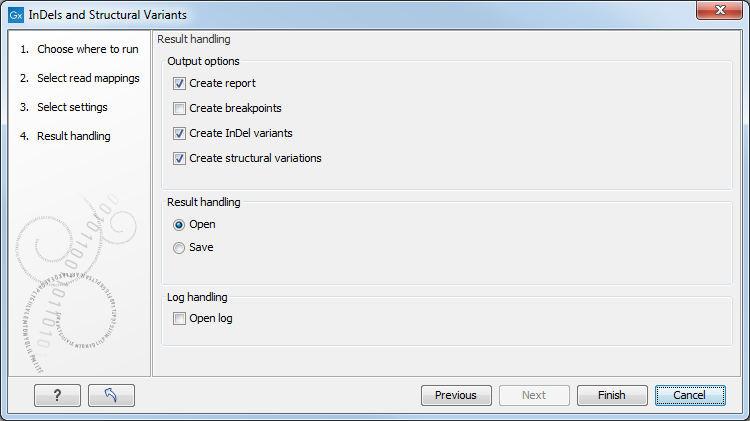
Figure 32.40: Select output formats.
- Create report When ticked, a report that summarizes information about the inferred breakpoints and variants is created.
- Create breakpoints When ticked, a track containing the detected breakpoints is created.
- Create InDel variants When ticked, a variant track containing the detected indels that fulfill the requirements for being 'variants' is created. These include:
- the detected insertions for which the allele sequence is inferred, but not those for which it is not known, or only partly known. As the algorithm relies on mapping two unaligned ends against each other for detecting insertions with inferred allele sequence, the maximum length of these that can potentially be detected depends on (1) the read length and (2) the "length fraction" parameter of the read mapper. With current read lengths and default settings you are unlikely to get insertions with inferred allele sequence larger than a couple of hundred, and hence will not see insertions in this track larger than that.
- medium sized deletions (those between six and 405 bp). All other deletions are put in the "Structural variants" track. The reason for not including all detected deletions in the indel track is that the main intended use of this track is to guide re-alignment. In our experience, the re-alignment algorithm performs best when only including the medium sized events. Notice that, in contrast to insertions, there is no upper limit on the length of deletions with inferred allele sequence that the algorithm can detect. This is because the allele sequence is trivial for deletions, whereas for insertions it must be inferred from the nucleotides in the unaligned ends.
- Create structural variations When ticked, a track containing the detected structural variants is created, including the insertions with unknown allele sequence and the deletions that are not included in the "InDel" track.
An example of the output from the InDels and Structural Variant tool is shown in figure 32.41. The output is described in detail in here.
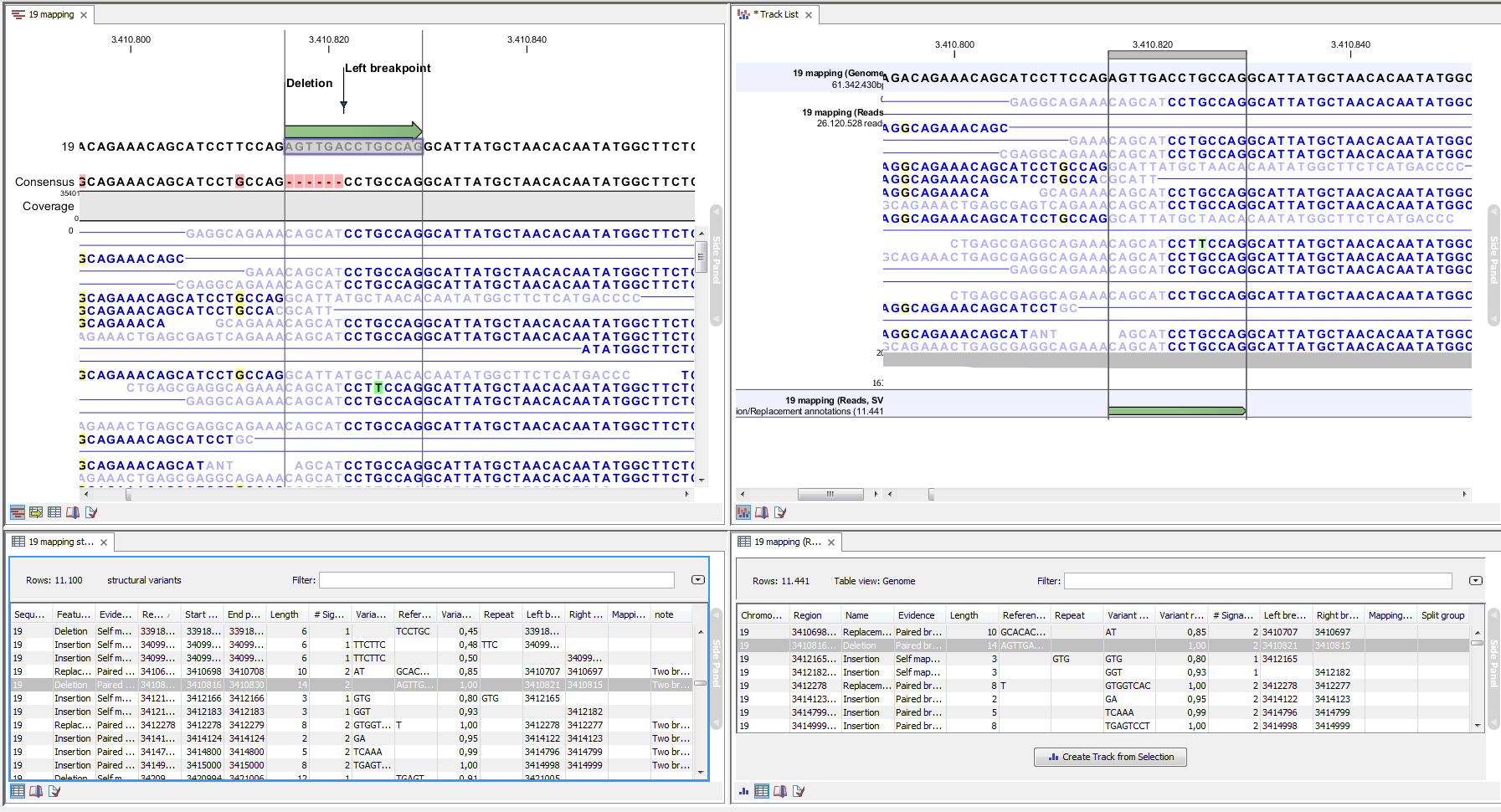
Figure 32.41: Example of the result of an analysis on a standalone read mapping (to the left) and on a reads track (to the right).