Illumina
CLC Genomics Workbench supports data from Illumina's Genome Analyzer, HiSeq 2000, NextSeq and the MiSeq systems.
To launch the Illumina importer, go to:
Import (![]() ) | Illumina (
) | Illumina (![]() ).
).
This opens a dialog where files can be selected and import options specified (figure 7.9).
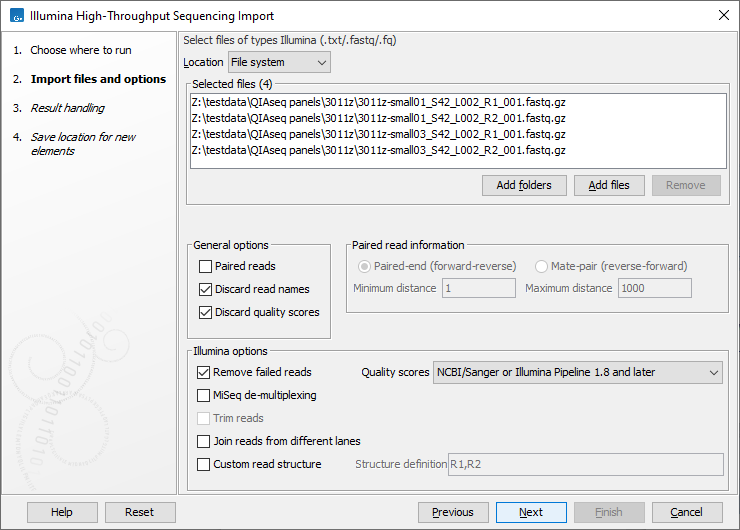
Figure 7.9: Importing data from Illumina systems.
Fastq (.fastq/.fq) files from Illumina can be imported. Uncompressed files as well as files compressed using gzip (.gz), zip (.zip) or bzip2 (.bz2) can be provided as input. The importer processes UMI information from the fastq read headers, see General notes on UMIs.
The drop down menu of input file locations includes the option BaseSpace. When selected, an Access BaseSpace... button is presented. Clicking this opens a browser window, where your Illumina BaseSpace credentials can be entered. After doing that, granting the CLC Workbench relevant access permissions and closing the browser window, you will be able to select files from BaseSpace in the Illumina High-Throughput Sequencing Import wizard. Your BaseSpace credentials remain valid for your current CLC Workbench session. BaseSpace configuration options are available in Preferences, see Advanced preferences.
The General options are:
- Paired reads. Files will be paired up based on their names, see Default rules for determining pairs of files below.
Under Paired read information:
- Choose the orientation of the paired reads, either Forward-reverse or Reverse-forward.
- Specify the insert sizes by setting Minimum distance and Maximum distance. Data sets with different insert sizes should be imported separately, with the correct minimum and maximum distance.
Read more about handling paired data in General notes on handling paired data.
- Discard read names. Read names can be discarded to save disk space without affecting analysis results. Keeping read names can be useful in some circumstances, such as when inspecting sequence list contents or when working downstream with subsets of sequences.
- Discard quality scores. Quality scores are visible in read mappings and are used by various tools, e.g. for variant detection. If quality scores are not relevant, use this option to discard them and reduce disk space and memory consumption.
Default rules for determining pairs of files
First, the selected files are sorted based on the file names. Sorting is alphanumeric, except for files coming off the CASAVA1.8 pipeline, where pairs are organized according to their identifier and chunk number.
For example, for files from CASAVA1.8, files with base names like: ID_R1_001, ID_R1_002, ID_R2_001, ID_R2_002, the files would be sorted in the order below, where it is assumed that files with names containing "R1" contain the first sequences of the pairs, and those containing "R2" in the name contain the second sequence of the pairs.
- ID_R1_001
- ID_R2_001
- ID_R1_002
- ID_R2_002
In this example, the data in files ID_R1_001 and ID_R2_001 are treated as a pair, and ID_R1_002, ID_R2_002 are treated as a pair.
The file names are then used to check if each prospective file pair in this sorted list is valid.
If the files in a pair seem to follow the following naming format:
<sample name>_L<at least one digit>_[R1|R2]_<at least one digit>,
then the files must contain the same sample name and lane information, in order to be valid.
If a prospective file pair does not follow this format, but the first file name does contain "_R1" and the second file name does contain "_R2", then the file pair is still considered valid. Note that if "_R1" or "_R2" occur more than once in a filename, the last occurrence in the name is used.
No data will be imported from file pairs that are not considered valid wrt. the above requirements. For such file pairs, a message will be printed in the log.
If the Join reads from different lanes option, in the Illumina options section of the dialog, is checked, then valid pairs of files with the same lane information in their file names will be imported into the same sequence list. If a valid pair of files do not contain the same lane information in their names, then no data is imported from those files and a message is printed in the log.
Within each file, the first read of a pair will have a 1 somewhere in the information line. In most cases, this will be a /1 at the end of the read name. In some cases though (e.g. CASAVA1.8), there will be a 1 elsewhere in the information line for each sequence. Similarly, the second read of a pair will have a 2 somewhere in the information line - either a /2 at the end of the read name, or a 2 elsewhere in the information line.
The organization of the files can be customized using the Custom read structure field, described in Illumina options below.
Illumina options
- Remove failed reads. Use this option to not import reads that did not pass a quality filter, as indicated within the fastq files.
Part of the header information for the quality score has a flag where Y means failed and N means passed. In this example, the read has not passed the quality filter:
@EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACG
If you import paired data and one read in a pair is removed during import, the remaining mate will be saved in a separate sequence list with single reads.
- MiSeq de-multiplexing. Using this option on MiSeq multiplexed data will divide reads into different files based on the "IndexSequence" of the read header:
@Instrument:RunID:FlowCellID:Lane:Tile:X:Y:UMI ReadNum:FilterFlag:0:IndexSequence
- Trim reads. When checked, reads are trimmed when a B is encountered at either end of the reads in the input file. This option is only available when the "Quality score" option has been set to Illumina Pipeline 1.5 to 1.7 as a B in the quality score has a special meaning as a trim clipping in this pipeline. This trimming is carried out whether or not you choose to discard quality scores during import.
- Join reads from different lanes.
When checked, fastq files from the same sequencing run but from different lanes are imported as a single sequence list.
Lane information is expected in the filenames as "_L<digits>", e.g. "L001" for lane 1. If this patterns occurs more than once in a filename, the last occurrence in the name is used. For example, for a filename "myFile_L001_L1.fastq" the lane information is assumed to be L1.
- Quality scores. There are four options for quality scores:
- NCBI/Sanger or Illumina 1.8 and later.
- Illumina Pipeline 1.2 and earlier.
- Illumina Pipeline 1.3 and 1.4.
- Illumina Pipeline 1.5 to 1.7.
- Custom read structure. If the default organization of Illumina files for import does not match what is needed, you can check custom read structure and specify the desired organization in the structure definition field. Fastq files are specified by the read information in the name (e.g. R1, R2, I1, I2). When separated by a space, the specified reads for a given spot are concatenated on import. When comma separated, a paired sequence list is imported, with the first sequence in the pair made up of the read or reads listed before the comma, and the second sequence made up of the read or reads listed after the comma.
For example:
- If
R2, R1was entered, a paired sequence list would be imported. The first sequence of each pair would contain a read from the R2 fastq file, and its partner would contain the corresponding read from the R1 fastq file. - If
I1 R1was entered, a sequence list containing single reads would be imported. Each read would contain sequence from the I1 fastq file prepended to sequence from the R1 fastq file. - If
R2 R1, R3was entered, a paired sequence list would be imported. The first sequence of each pair would contain a read from the R2 fastq file prepended to the corresponding read from the R1 fastq file. The second sequence of each pair would contain the corresponding read from the R3 fastq file.This could represent the situation where R1 contains forward reads, R3 has reverse reads, and R2 contains molecular indices.
- If
In the next wizard step, options are presented for how to handle the results. If you choose to Save the results, an option called "Create subfolders per batch unit" becomes available. When that option is checked, each sequence list is saved into a separate folder under the location selected to save results to. This can be useful for organizing subsequent analysis results and for batch processing.
Subsections