Over-representation analyses
Please note that if the coverage is below 0.005% across the end positions of the reads, then these positions will not be shown in the enriched 5-mer distribution plot described below (see section 29.1).
- Enriched 5-mer distribution
- The 5-mer analysis examines the enrichment of penta-nucleotides. The enrichment of 5-mers is calculated as the ratio of observed and expected 5-mer frequencies. The expected frequency is calculated as product of the empirical nucleotide probabilities that make up the 5-mer. (Example: given the 5-mer = CCCCC and cytosines have been observed to 20% in the examined sequences, the 5-mer expectation is
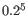 ). Note that 5-mers that contain ambiguous bases (anything different from A/T/C/G) are ignored. This analysis calculates the absolute coverage and enrichment for each 5-mer (observed/expected based on background distribution of nucleotides) for each base position, and plots position vs enrichment data for the top five enriched 5-mers (or fewer if less than five enriched 5-mers are present). It will reveal if there is a bias at certain positions along the read length. This may originate from non-trimmed adapter sequences, poly A tails and more.
). Note that 5-mers that contain ambiguous bases (anything different from A/T/C/G) are ignored. This analysis calculates the absolute coverage and enrichment for each 5-mer (observed/expected based on background distribution of nucleotides) for each base position, and plots position vs enrichment data for the top five enriched 5-mers (or fewer if less than five enriched 5-mers are present). It will reveal if there is a bias at certain positions along the read length. This may originate from non-trimmed adapter sequences, poly A tails and more.
- Sequence duplication levels
- The duplicated sequences analysis identifies sequence reads that have been sequenced multiple times. A high level of duplication may indicate an enrichment bias, as for instance introduced by PCR amplification. Please note that multiple input sequence lists will be considered as one federated data set for this analysis. Batch mode can be used to generate separate reports for individual sequence lists.
In order to identify duplicate reads the tool examines all reads in the input and uses a clone dictionary containing per clone the read representing the clone and a counter representing the size of the clone. For each input read these steps are followed: (1) check whether the read is already in the dictionary. (2a) if yes, increment the according counter and continue with next read. (2b) if not, put the read in the dictionary and set its counter to 1.
To achieve reasonable performance, the dictionary has a maximum capacity of 250,000 clones. To this end, step 2a involves a random decision as to whether a read is granted entry into the clone dictionary. Every read that is not already in the dictionary has the same chance T of entering the clone dictionary with T = 250,000 / total amount of input reads. This design has the following properties:
- The clone dictionary will ultimately contain at most 250,000 entries.
- The sum of all clone sizes in the dictionary amounts at most to the total number of input reads.
- Because of T being constant for all input reads, even a cluster of reads belonging to the same clone and first occurring towards the end of the input can be detected.
- Because of the random sampling, the tool might underestimate the size of a read clone, specifically if its first read representative does not make it into the dictionary. The ratio is that a larger clone has a higher cumulative chance of being eventually represented in the dictionary than a smaller clone.
Because all current sequencing techniques tend to report decreasing quality scores for the 3' ends of sequences, there is a risk that duplicates are NOT detected, merely because of sequencing errors towards their 3' ends. The identity of two sequence reads is therefore determined based on the identity of the first 50nt from the 5' end.
The results of this analysis are presented in a plot and a corresponding table correlating the clone size (duplication count) with the number of clones of that size. For example, if the input contains 10 sequences and each sequence was seen exactly once, then the table will contain only one row with duplication-count=1 and sequence-count=10. Note: due to space restrictions the corresponding bar-plot shows only bars for duplication-counts of x=[0-100]. Bar-heights of duplication-counts >100 are accumulated at x=100. Please refer to the table-report for a full list of individual duplication-counts.
- Duplicated sequences
- This results in a list of actual sequences most prevalently observed. The list contains a maximum of 25 (most frequently observed) sequences and is only present in the supplementary report.