General filters
The General filters relate to the regions and reads in the read mappings that should be considered, and the amount of evidence the user wants to require for a variant to be called (figure 32.8):
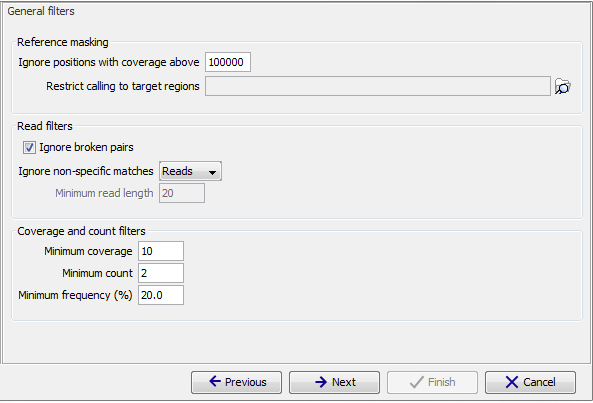
Figure 32.8: General filters. The values shown are those that are default for Fixed Ploidy Variant detection.
Note on the use of the Low Frequency Variant Detection tool with Whole Genome Sequencing data: The default settings for the Low Frequency Variant Detection tool are optimized for targeted resequencing protocols, and NOT whole genome sequencing (e.g. cancer gene panels) where it is not uncommon to have modest coverage for most part of the mapping, and abnormal areas (typically repeats around the centromeres) with very high coverage. Looking for low frequency variants in high coverage areas will exhaust the machine memory because there will be many low frequency variants due to some reads originating from near identical repeat sequences or simple sequencing errors.In order to run the tool on WGS data the parameter Ignore positions with coverage above should be adjusted to a lower number (typically 1000).
Reference masking
The 'Reference masking' filters allow the user to only perform variant calling (including error model estimation) in specific regions. In addition to selecting an annotation track, there are two parameters to specify:
- Ignore positions with coverage above: All positions with coverage above this value will be ignored when inspecting the read mapping for variants. The option is highly useful in cases where you have a read mapping which has areas of extremely high coverage as are areas around centromeres in whole genome sequencing applications for example.
- Restrict calling to target regions: Only positions in the regions specified will be inspected for variants. However, note that insertions situated directly to the right of a target region will also be included in the variant track because their reference allele is included inside the target.
Read filters
The Read filters determine which reads (or regions) should be considered when calling the variants.- Ignore broken pairs: When ticked, reads from broken pairs are ignored. Broken pairs may arise for a number of reasons, one being erroneous mapping of the reads. In general, variants based on broken pair reads are likely to be less reliable, so ignoring them may reduce the number of spurious variants called. However, broken pairs may also arise for biological reasons (e.g. due to structural variants) and if they are ignored some true variants may go undetected. Please note that ignored broken pair reads will not be considered for any non-specific match filters.
- Non-specific match filter: Non-specific matches are likely to come from repeat region whose exact mapping location is uncertain. In general, variants based on non-specific matches are likely to be less reliable. However, as there are regions in the genome that are entirely perfect repeats, ignoring non-specific matches may have the effect that true variants go undetected in these regions.
There are three options for specifying to which 'extent' the non-specific matches should be ignored:
- 'No': they are not ignored.
- 'Reads': they are ignored.
- 'Region': when this option is chosen no variants are called in regions covered by at least one non-specific match. In this case, the minimum length of reads that are allowed to trigger this effect has to be stated, as really short reads will usually be non-specific even if they do not stem from repeat regions.
Coverage and count filters
These filters specify absolute requirements for the variants to be called. Note that suitable values for these filters are highly dependent on the coverage in the sample being analyzed:- Minimum coverage: Only variants in regions covered by at least this many reads are called.
- Minimum count: Only variants that are present in at least this many reads are called.
- Minimum frequency: Only variants that are present at least at the specified frequency (calculated as 'count'/'coverage') are called.