Troubleshooting SRA downloads
This section covers various issues that may be observed when using the Download Reads and Metadata functionality, available after a search in SRA has been run using Search for Reads in SRA.
Reads cannot be downloaded for certain SRA runs
If reads cannot be downloaded for certain SRA runs, the runs are listed in a Problems panel together with a description of the problem. It is usually still possible to download reads for the remaining runs.
The most common problems are:
- "The selected SRA reads contain no spots, and cannot be imported in the workbench.": The run has no associated sequencing data.
- "The selected SRA reads are dbGaP restricted.": For data protection reasons, you must request access to these reads. Requests and download cannot happen within the workbench, but you can follow the procedures here: https://www.ncbi.nlm.nih.gov/books/NBK5295/.
- "The selected SRA reads are made with an unsupported sequencing platform.": For example, Complete Genomics reads consist of eight regions separated by gaps of variable lengths, and should be analyzed by specialist tools.
No values in the Biological reads or Technical reads columns of the results table
If there are no values in the Biological reads or Technical reads columns, the SRA entry may contain inconsistent information in an SRA entry. Downloading such entries is usually possible, but what is downloaded will depend on the circumstances.
For example, if a run has more than one read per spot, but is not marked as paired, the Biological reads and Technical reads columns will be blank. Downloading this data will result in a sequence list containing single reads from R1. This would be fine in a case like SRR16530746, where R1 contains the read information (and R2 contains no bases). However, it may not be fine for other entries.
In such cases, we recommend checking the imported sequence list contains the expected data.
Paired end settings table issues
- "?" in Reads Available column and "Use SRA Defaults" in Import Read Structure column
When there are more than 2 reads available for a run marked as paired, but no information about the read structure is available, "(?)" appears beside each read set in the "Reads Available" column, and "Use SRA Defaults" appears in the Import Read Structure column. If not configured further, reads that SRA defines as biological by default are imported.
In many cases, this is fine, but we recommend explicitly defining the read structure. If you do not, please check the resulting sequence lists to ensure the expected data is present. Click on a run accession in the results table to go directly to the SRA webpage about that run.
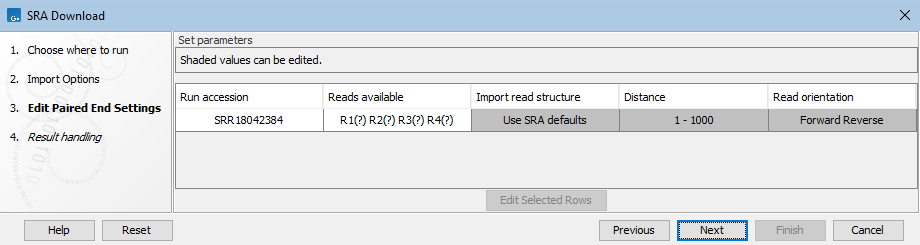
Figure 11.13: This run contains more than 2 reads per spot but no explicit read structure information. Unless configured further, reads will be imported according to SRA default handling, with only reads SRA interprets as biological being imported. - "Use SRA defaults" warning under Import read structure column
In the "Edit Paired End Settings" step of the SRA Download wizard, text in the "Import read structure" column will state "Use SRA defaults" and include a warning exclamation mark if the nature of the reads in SRA does not match what is expected. For example, a run is not expected to have three or more biological reads. (figure 11.14).
If you choose to import data from such runs, we recommend the read structure is explicitly defined, as described in Downloading reads and metadata from SRA. If it is not, biological reads will be imported using default settings. This may or may not result in a sequence list containing data relevant for downstream processing.
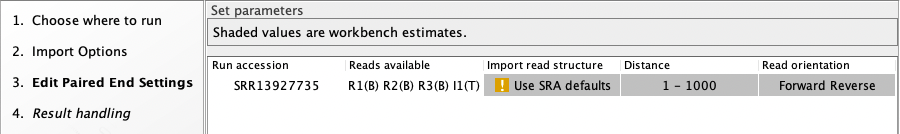
Figure 11.14: his run has 3 biological reads, which is not expected. A warning icon is shown in the Import read structure column. If the data needs to be downloaded, then explicitly defining the read structure in a case like this is recommended.