Annotate with GFF/GTF/GVF file
Annotate with GFF/GTF/GVF files adds annotations from GFF3, GTF, or GVF files to DNA, RNA, or protein sequences. Annotations can be added to sequences stored individually (![]() ) (
) (![]() ) (
) (![]() ) or in lists (
) or in lists (![]() ) (
) (![]() ). The names in the first column in the file must match the names of the sequences to be annotated. If this is not the case, either the names in the annotation file, or the names of the sequences, must be updated.
). The names in the first column in the file must match the names of the sequences to be annotated. If this is not the case, either the names in the annotation file, or the names of the sequences, must be updated.
Tools are available for renaming sequences or sequences in sequence lists:
Importing standard reference data
Before proceeding to use Annotate with GFF/GTF/GVF file, please refer to References management. This covers how to download standard reference data, including annotations, provided by QIAGEN and other public sources using the Reference Data Manager, which is part of the Workbench. If the reference data you are interested in is available through the Reference Data Manager, it is usually easier to use that rather than taking the steps outlined in this section.
If the reference data you need is not available through the Reference Data Manager, and you are working with track-based data, please refer to Import tracks, rather than using Annotate with GFF/GTF/GVF file.
How annotations are applied
Annotations from each line in the annotation file are placed on the sequence with the name given in the first column. Special treatment is given to annotations of the types CDS, exon, mRNA, transcript and gene. For these, the following applies:
- A gene annotation is generated for each gene_id. The region annotated extends from the leftmost to the rightmost positions of all annotations that have the gene_id (gtf-style).
- CDS annotations that have the same transcriptID are joined to one CDS annotation (gtf-style). Similarly, CDS annotations that have the same parent are joined to one CDS annotation (gff-style).
- If there is more than one exon annotation with the same transcriptID these are joined to one mRNA annotation. If there is only one exon annotation with a particular transcriptID, and no CDS with this transcriptID, a transcript annotation is added instead of the exon annotation (gtf-style).
- Exon annotations that have the same mRNA as parent are joined to one mRNA annotation. Similarly, exon annotations that have the same transcript as parent, are joined to one transcript annotation (gff-style).
Note that genes and transcripts are linked by name only (not by position, ID etc).
Running the tool
To run the Annotate with GFF/GTF/GVF file tool, go to:
Tools| Classical Sequence Analysis (![]() )| Classical Sequence Analysis (
)| Classical Sequence Analysis (![]() ) | General Sequence Analysis (
) | General Sequence Analysis (![]() )| Annotate with GFF/GTF/GVF file (
)| Annotate with GFF/GTF/GVF file (![]() )
)
After selecting the sequence to annotate, the next step will look like that shown in figure 18.1.
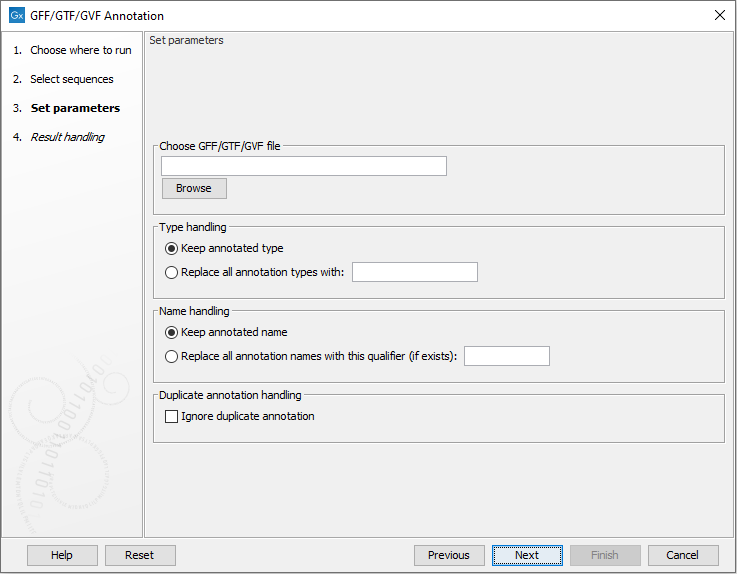
Figure 18.1: Select a GFF, GTF or GVR file by clicking on the Browse button.
Click on Browse to select a GFF, GTF or GVF file. After working through handling options, described below, your sequences will be annotated by the information from that file.
Name handling
Annotations are named in the following, prioritized way:
- If one of the following qualifiers are present, it will be used for naming (prioritized):
- Name
- Gene_name
- Gene_ID
- Locus_tag
- ID
- If none of these are found, the annotation type will be used as name.
If you provide a qualiifer, it must be written identically to the corresponding qualifier name in the annotation file.
Transcript annotations are handled separately, since they inherit the name from the gene annotation.
Figure 18.2: You can choose Replace all annotation names with the specified qualifier.
Type handling
You can overrule feature types in the annotation file by choosing Replace all annotation types with and specifying a type to use.
Ignore duplicate annotation
When the Ignore duplicate annotation option is checked, only one instance of duplicate annotations will be added to the sequence.
Create log
In the Result handling section of the wizard, check the Create log box results to create a log that includes information like the number of annotations found and if there are any that are could not be placed on the sequence. This information can help with troubleshooting when annotations are not added to a sequence when they were expected to be.