SAM, BAM and CRAM mapping files
The CLC Genomics Workbench supports import of files in the following alignment map formats for storing large nucleotide sequence alignments:
- SAM (Sequence Alignment/Map)
- BAM (Binary Alignment/Map)
- CRAM (Compressed Reference-oriented Alignment/Map)
See https://samtools.github.io/hts-specs/ for specifications for these formats.
Alignments from a SAM/BAM/CRAM file can be imported as a reads track or as stand-alone read mappings.
To import the reads in a SAM/BAM file as a sequence list, disregarding any alignment information, please use the Standard Import instead (see Standard import).
Note that importing SAM/BAM/CRAM files can be very memory-consuming for large data sets, and particularly those with many reads marked as members of broken pairs in the mapping.
To launch the SAM/BAM/CRAM importer, go to:
Import (![]() ) | SAM/BAM/CRAM Mapping Files (
) | SAM/BAM/CRAM Mapping Files (![]() ).
).
This opens a dialog where files can be selected and import options specified (figure 7.23).
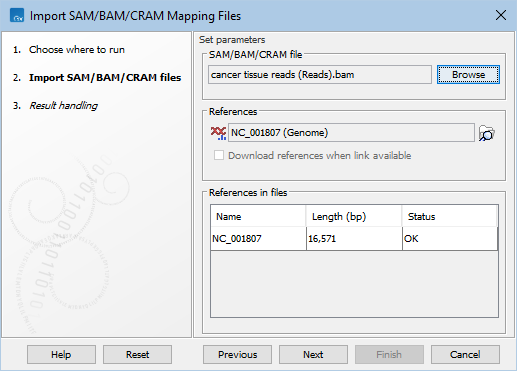
Figure 7.23: Choosing SAM/BAM/CRAM file and reference(s).
Providing references for the SAM/BAM/CRAM file
The reference sequence(s) that are referred to within the SAM/BAM/CRAM file must be specified in the 'Set parameters' wizard step (figure 7.23):
- If the SAM/BAM/CRAM file already contains information about where to find the reference(s), tick Download references when link available.
- If the input reference(s) are present in the CLC Genomics Workbench, click on the "Find in folder" icon (
 ) to select the reference(s).
) to select the reference(s).
Occurrences of disallowed characters according to the specification at https://samtools.github.io/hts-specs/SAMv1.pdf (whitespaces \ , " ` ' @ ( ) [ ] < >) in the input references are replaced by _ (underscore). Additionally, = and * are only disallowed at the beginning of the reference names. E.g., an input reference named
*my=reference@sequenceis considered the same as the reference_my=reference_sequencereferred to within the SAM/BAM/CRAM file.When importing SAM or BAM files, a range of different case-insensitive synonyms are used when matching input reference names with those specified in the file, e.g.:
- Chromosome 1: 1, chr1, chromosome_1, nc_000001
- Chromosome M: m, mt, chrm, chrmt, chromosome_m, chromosome_mt, nc_001807
- Chromosome Y: y, chry, chromosome_y, nc_000024
The table under 'References in files' contains the references that are referred to within the SAM/BAM/CRAM file, with their name, length, and a status. The status indicates whether a given reference referred to within the SAM/BAM/CRAM file is present in the input references. The status can be:
- OK. There is a reference in the input references with this name and length.
- Length differs. There is a reference in the input references with this name, but with a different length.
- Download link available. The SAM/BAM/CRAM file contains a URL for this reference. Tick Download references when link available to automatically download the reference.
- Will download. The SAM/BAM/CRAM file contains a URL for this reference and Download references when link available is already ticked. The reference is automatically downloaded.
- Missing, download link not available. There is no reference in the input references with this name, and there is no URL available in the SAM/BAM/CRAM file for downloading the reference.
A reference is 'matched' when the status is either OK or Will download. Only reads mapping to a matched reference are imported from SAM and BAM files. Import of CRAM files fails when there are unmatched references.
For references located on a CLC Genomics Server, the table is empty. The importer can be launched, regardless of whether the correct references are selected, but it leads to an error in cases where they are not.
Output options
In the 'Result handling' wizard step, the output options for the importer can be configured (figure 7.24):
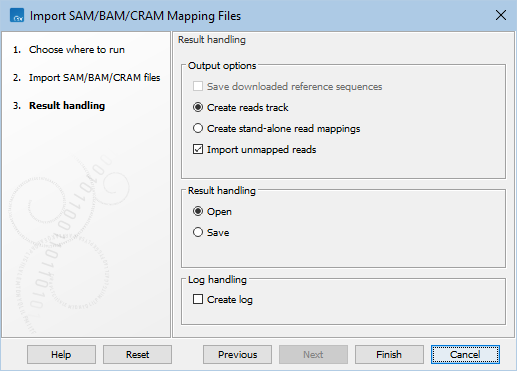
- Save downloaded reference sequences. This option is enabled if the option Download references when link available is selected in the 'Set parameters' wizard step.
- Create reads track. Mapped reads are imported as a reads track.
- Create stand-alone read mappings. Mapped reads are imported as stand-alone read mappings. When there is only one reference, the result is a single read mapping (
 ), otherwise the result is a multi-mapping element (
), otherwise the result is a multi-mapping element ( ).
).
- Import unmapped reads. Unmapped reads are imported into sequence lists. One sequence list is created per read group.
- If unmapped reads are part of an intact pair, they are imported into a sequence list of paired data.
- If unmapped reads are single reads or a member of a pair that did not map while its mate did, they are imported into a sequence list containing single reads.
Only reads mapping to a matched reference are imported from SAM and BAM files. Import of CRAM files fails when there are unmatched references.
For files containing multiple alignment records for a single read, only the primary alignment (see https://samtools.github.io/hts-specs/SAMv1.pdf) is imported.