Demultiplex Reads
Multiplexing techniques are often used when sequencing different samples in one sequencing run. One method used is to tag the sequences with a unique identifier during the preparation of the sample for sequencing [Meyer et al., 2007].
With this technique, each sequence read will have a sample-specific tag, which is a specific sequence of nucleotides before or after the sequence of interest, see figure 29.17.
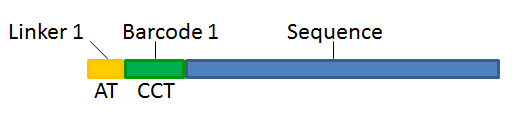
Figure 29.17: Tagging the target sequence, which in this case is single reads from one sample.
Demultiplex Reads looks for matches between reads and these sample-specific tags, also called barcodes or indexes, to group the reads by sample. The reads for each sample can then be used in downstream analyses to generate sample-specific results.
When Demultiplex Reads is used within a workflow, the 'Demultiplexed Reads' output channel needs to be connected to an Iterate element. The sets of reads to be analyzed together, i.e. the batch units (see Running part of a workflow multiple times), are determined by the barcodes. See Running Demultiplex Reads in workflows for further details.
Demultiplexing is often carried out on the sequencing machine so that the sequencing reads are already separated according to sample before importing it into the CLC Genomics Workbench. This is often the best option, if available.
Subsections