Deriving the posterior probabilities of the site types
We will call a variant at a site if the sum of the posterior probabilities of the non-homozygous reference site types is larger than the user-specified cut-off value. For this we need to be able to calculate the posterior site type probabilities. We here derive the formula for these.
Using the Bayesian approach we can write the posterior probability of a site type, ![]() , as follows:
, as follows:
where
To derive the likelihood of the data,
![]() , we first need some notation: For a given site type,
, we first need some notation: For a given site type, ![]() , let
, let ![]() be the probability that an allele from this site type has the nucleotide
be the probability that an allele from this site type has the nucleotide ![]() . The
. The ![]() probabilities are known and are determined by the ploidy: For a diploid organism,
probabilities are known and are determined by the ploidy: For a diploid organism, ![]() if
if ![]() is a homozygous site and
is a homozygous site and ![]() is one of the alleles in
is one of the alleles in ![]() , whereas it is 0.5 if
, whereas it is 0.5 if ![]() is a heterozygous and
is a heterozygous and ![]() is one of the alleles in
is one of the alleles in ![]() , and it is 0, if
, and it is 0, if ![]() is not one of the alleles in
is not one of the alleles in ![]() . For a triploid organism, the
. For a triploid organism, the ![]() will be either 0, 1/3, 2/3 or 1.
will be either 0, 1/3, 2/3 or 1.
With this definition, we can write the likelihood of the data
![]() in a site
in a site ![]() as:
as:
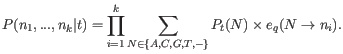
Inserting this expression for the likelihood, and the prior site type frequencies ![]() and
and ![]() for
for ![]() and
and ![]() , in the expression for the posterior probability (31.1), we thus have the following equation for the posterior probabilities of the site types:
, in the expression for the posterior probability (31.1), we thus have the following equation for the posterior probabilities of the site types:
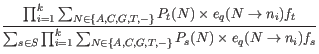
The unknowns in this equation are the prior site type probabilities,
![]() , and the error rates
, and the error rates
![]() . Once these have been estimated, we can calculate the posterior site type probabilities using the equation 31.3 for each site type, and hence, for each site, evaluate whether the sum of the posterior probabilities of the non-homozygous reference site types is larger than the cut-off. If so, we will set out current estimated site type to be that with the highest posterior probability.
. Once these have been estimated, we can calculate the posterior site type probabilities using the equation 31.3 for each site type, and hence, for each site, evaluate whether the sum of the posterior probabilities of the non-homozygous reference site types is larger than the cut-off. If so, we will set out current estimated site type to be that with the highest posterior probability.