Significance thresholds
Clicking Next will display the dialog shown in figure 26.14.
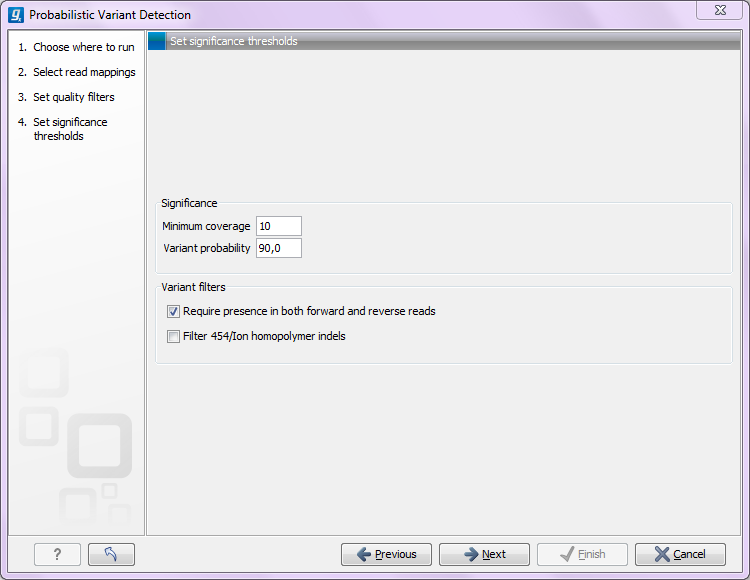
Figure 26.14: Significance thresholds.
The follow parameters can be set:
- Minimum coverage The minimum number of reads aligned to the site to be considered a potential variant.
- Variant probability This is the posterior probability from the Bayesian approach.
Below the significance settings, there are two filters that can be useful for removing false positives:
- Require presence in both forward and reverse reads. Some systematic sequencing errors can be triggered by a certain combination of bases. This means that sequencing one strand may lead to sequencing errors that are not seen when sequencing the other strand (see [Nguyen et al., 2011] for a recent study with Illumina data). This can easily lead to false positive variant calls, and by checking this filter, the minimum ratio between forward and reverse reads supporting the variant should be at least 0.05. In this way, systematic sequencing errors of this kind can be eliminated. The forward/reverse reads balance is also reported for each variant in the result (see Variant data).
- Filter 454/Ion homopolymer indels. The 454 and Ion Torrent/Proton sequencing platforms exhibit weaknesses when determining the correct number of the same kind of nucleotides in a homopolymer region (e.g. AAA). This leads to a high false positive rate for calling InDels in these regions. This filter is very basic: it removes all indels that are found within or just next to a homopolymer region. A homopolymer region is defined as at least two consecutive identical bases in the reference.