De novo assembly parameters
To start the assembly:
Toolbox | De Novo Sequencing (![]() ) | De Novo Assembly (
) | De Novo Assembly (![]() )
)
In this dialog, you can select one or more sequence lists or single sequences.
Click Next to set the parameters for the assembly. This will show a dialog similar to the one in figure 28.18.
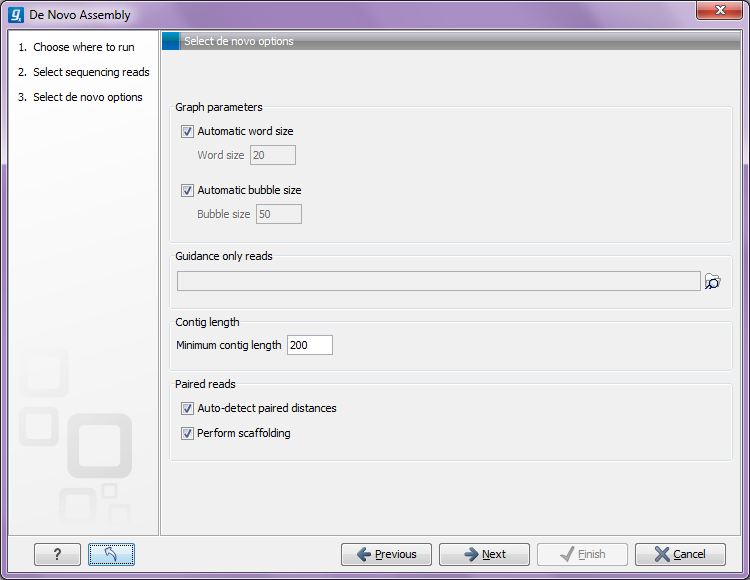
Figure 28.18: Setting parameters for the assembly.
At the top, you select the Word size and the Bubble size to be used. The principles of setting the word size are described in How it works. When using automatic calculation, you can see the word size in the History (![]() ) of the result files. Please note that the range of word sizes is 12-24 on 32-bit computers and 12-64 on 64-bit computers. The meaning of the bubble size parameter is explained in Bubble resolution. The bubble size used when the setting is automatic is 50 for reads shorter than 110 bp, and for longer reads it is the average read length. The value used is also recorded in the History (
) of the result files. Please note that the range of word sizes is 12-24 on 32-bit computers and 12-64 on 64-bit computers. The meaning of the bubble size parameter is explained in Bubble resolution. The bubble size used when the setting is automatic is 50 for reads shorter than 110 bp, and for longer reads it is the average read length. The value used is also recorded in the History (![]() ) of the result files.
) of the result files.
The next option is to specify Guidance only reads. The reads supplied here will not be used to create the de Bruijn graph and subsequent contig sequence but only used to resolved ambiguities in the graph (see Resolve repeats using reads and Optimization of the graph using paired reads). With mixed data sets from different sequencing platforms, we recommend using sequencing data with low error rates as the main input for the assembly, whereas data with more errors should be specified only as Guidance only reads. This would typically be long reads or paired data sets.
You can also specify the Minimum contig length when doing de novo assembly. Contigs below this length will not be reported. The default value is 200 bp. For very large assemblies, the number of contigs can be huge (over a million), in which case the data structures when mapping reads back to contigs will be very large and take a very long time to handle. In this case, it is a great advantage to raise the minimum contig length to reduce the number of contigs that have to be incorporated into this data structure.
At the bottom, there is an option to Perform scaffolding. The scaffolding step is explained in greater detail in Optimization of the graph using paired reads. This will also cause scaffolding annotations to be added to the contig sequences (except when you also choose to Update contigs, see below).
Finally, there is an option to Auto-detect paired distances. This will determine the paired distance (insert size) of paired data sets. If several paired sequence lists are used as input, a separate calculation is done for each one to allow for different libraries in the same run. The History (![]() ) view of the result will list the distance used for each data set.
) view of the result will list the distance used for each data set.
If the automatic detection of pairs is not checked, the assembler will use the information about minimum and maximum distance recorded on the input sequence lists (see General notes on handling paired data).
For mate-pair data sets with large insert sizes, it may not be possible to infer the correct paired distance. In this case, the automatic distance calculation should not be used.
The best way of checking this is to run a read mapping using the contigs from the de novo assembly as reference and the mate-pair library as reads, and then check the Detailed mapping report. There is a paired distance distribution graph that can be used to check whether the distance estimated by the assembler fits in the distribution found in the read mapping.
When you click Next, you will see the dialog shown in figure 28.19
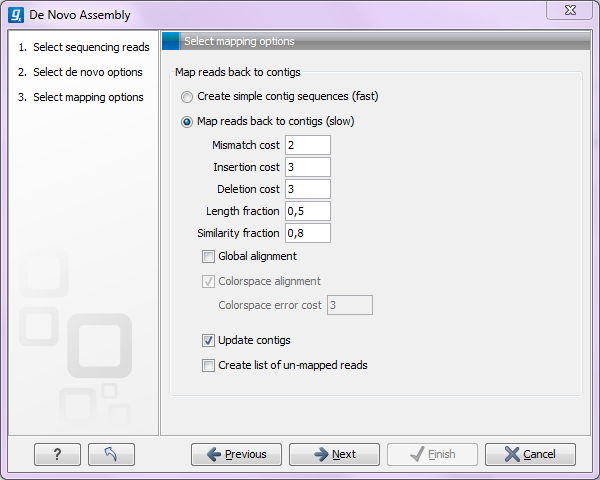
Figure 28.19: Parameters for mapping reads back to the contigs.
At the top, you choose whether a read mapping should be performed after the initial contig creation. If you choose to do that, you can specify the parameters for the read mapping. These are all explained in The read mapper tool.
At the bottom, you can choose to Update contigs based on mapped reads. This means that the original contig sequences produced from the de novo assembly will be updated to reflect the mapping of the reads (in most cases it will mean no change, but in some cases, the subsequent mapping step leads to new information). In effect, this means that all contig sequences in the output will be supported by at least one read mapped back. Note that if this option is selected, the contig lengths may get below the threshold specified in figure 28.18 because this threshold is applied to the original contig sequences. If the Update contigs based on mapped reads option is not selected, the original contig sequences from the assembler will be preserved completely also in situations where the reads that are mapped back do not support the contig sequences.