Calculation of sequence logos
A comprehensive walk-through of the calculation of the information
content in sequence logos is beyond the scope of this document but
can be found in the original paper by [Schneider and Stephens, 1990].
Nevertheless, the conservation of every position is defined as
![]() which is the difference between the maximal entropy
(
which is the difference between the maximal entropy
(![]() ) and the observed entropy for the residue distribution
(
) and the observed entropy for the residue distribution
(![]() ),
),
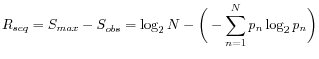
![]() is the observed frequency of a amino acid residue or
nucleotide of symbol
is the observed frequency of a amino acid residue or
nucleotide of symbol ![]() at a particular position and
at a particular position and ![]() is the number of distinct symbols for the sequence alphabet, either 20 for proteins or four for DNA/RNA. This means that the maximal sequence information content per position is
is the number of distinct symbols for the sequence alphabet, either 20 for proteins or four for DNA/RNA. This means that the maximal sequence information content per position is
![]() for DNA/RNA and
for DNA/RNA and
![]() for proteins.
for proteins.
The original implementation by Schneider does not handle sequence gaps.
We have slightly modified the algorithm so an estimated logo is presented in areas with sequence gaps.
If amino acid residues or nucleotides of one sequence are found in an area containing gaps, we have chosen to show the particular residue as the fraction of the sequences. Example; if one position in the alignment contain 9 gaps and only one alanine (A) the A represented in the logo has a hight of 0.1.
Other useful resources
The website of Tom Schneider
http://www-lmmb.ncifcrf.gov/~toms/
WebLogo
http://weblogo.berkeley.edu/