Import tracks
Tracks (see Tracks) are imported in a special way, because extra information is needed in order to interpret the files correctly.
Tracks are imported using:
click Import (![]() ) in the Toolbar | Tracks
) in the Toolbar | Tracks
This will open a dialog as shown in figure 6.16.
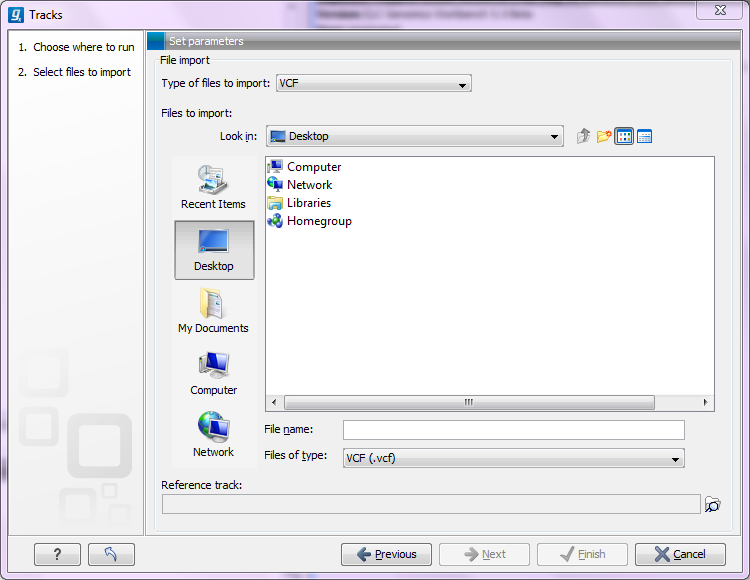
Figure 6.16: Define the reference genome.
At the top, you select the file type to import. Below, select the files to import. The formats currently accepted are:
- FASTA
- This is the standard fasta importer that will produce a sequence track rather than a standard fasta sequence. Please note that this could also be achieved by importing using Standard import and subsequently converting the sequence or sequence list to a track (see Converting data to tracks and back).
- GFF/GTF/GVF
- Annotations in gff/gtf/gvf formats. This is explained in detail in the user manual for the GFF annotation plug-in: www.clcbio.com/clc-plugin/annotate-sequence-with-gff-file/. In the context of the Genomics Gateway, this can be particular useful for downloading gene and transcript annotations in gtf format and variant data in gvf format from Ensembl: http://www.ensembl.org/info/data/ftp/index.html.
- VCF
- This is the file format used for variants by the 1000 Genomes Project and it has become a standard format. Read how to access data at http://www.1000genomes.org/data#DataAccess.
- BED
- Simple format for annotations. Read more at http://genome.ucsc.edu/FAQ/FAQformat.html#format1. This format is typically used for very simple annotations, for example target regions for sequence capture methods.
- Complete Genomics master var file
- This is the file format used by Complete Genomics for all kinds of variant data and can be used to analyze and visualize the variant calls made by Complete Genomics. Please note that you can import evidence files with the read alignments into the CLC Genomics Workbench as well (refer to the Complete Genomics import section of the Workbench user manual).
- Wiggle
- The Wiggle format as defined by UCSC (http://genome.ucsc.edu/goldenPath/help/wiggle.html), is used to hold continuous data like conservation scores, GC content etc. When imported into the CLC Genomics Workbench, a graph track is created. An example of a popular Wiggle file is the conservation scores from UCSC which can be download for human from http://hgdownload.cse.ucsc.edu/goldenPath/hg19/phastCons46way/.
- UCSC variant database table dump
- This is mainly intended to allow you to import the popular Common SNPs variant set from UCSC. The file can be downloaded from the UCSC web site here: http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/snp132Common.txt.gz. Other sets of variant annotation can also be downloaded in this format. The files ending with
.txt.gzon this list can be used: http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/.
For all of the above, zip files are also supported.
Please note that for human data, there is a difference between the UCSC genome build and Ensembl/NCBI for the mitochondrial genome. This means that for the mitochondrial genome, data from UCSC should not be mixed with data from other sources (see http://genome.ucsc.edu/cgi-bin/hgGateway?db=hg19).
Most of the data above is annotation data and if the file includes information about allele variants (like VCF, Complete Genomics and GVF), it will be combined into one variant track that can be used for finding known variants in your experimental data. When the data cannot be recognized as variant data, one track is created for each annotation type.
For all types of files except fasta, you need to select a reference track as well. This is because most the annotation files do not contain enough information about chromosome names and lengths which are necessary to create the appropriate data structures.