Find broken pair mates
Figure 25.21 shows an example of a read mapping with paired reads (shown in blue). In this particular region, there are some broken pairs (red and green reads). Pairs are marked as broken if the respective orientation or distance between the reads is not right (see general info on handling paired data), or if one of the reads do not map at all.
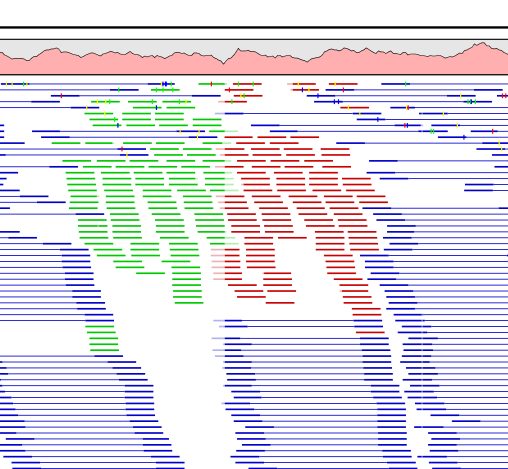
In some situations it is useful to investigate where the mate of the broken pairs map. This would indicate genomic rearrangements, mis-assemblies of de novo assembly etc. In order to see this, select the region in question on the reference sequence, right-click and choose Find Broken Pair Mates.
This will open the dialog shown in figure 25.22.
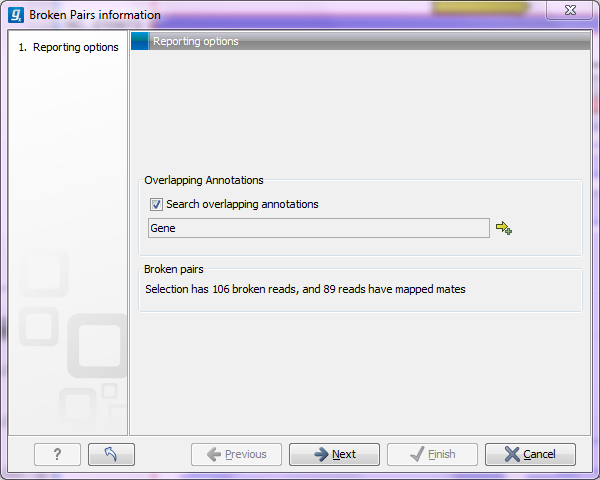
Figure 25.22: Finding the mates of broken pairs.
The purpose of this dialog is to let you specify if you want to annotate the resulting broken pair overview with annotation information. In this case, you would see if there are any overlapping genes at the position of the mates.
In addition, the dialog provides an overview of the broken pairs that are contained in the selection.
Click Next and Finish, and you will see an overview table as shown in figure 25.23.
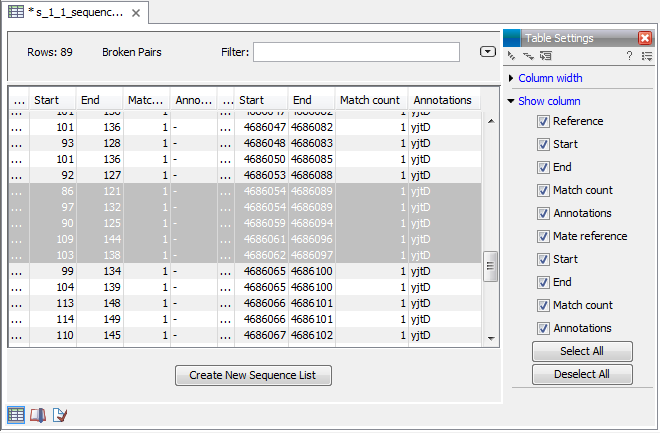
Figure 25.23: An overview of the broken pairs.
The table includes the following information for both parts of the pair:
- Reference
- The name of the reference sequence where it is mapped
- Start and end
- The position on the reference sequence where the read is aligned
- Match count
- The number of possible matches for the read. This value is always 1, unless the read is a non-specific match (marked in yellow)
- Annotations
- Shows a list of the overlapping annotations, based on the annotation type selected in figure 25.22.
You can select some or all of these broken pairs and extract them as a sequence list for further analysis by clicking the Create New Sequence List button at the bottom of the view.