Finding the right reference sequence for RNA-Seq
For prokaryotes, the reference sequence needed for RNA-Seq is quite simple. Either you input a genome annotated with gene annotations, or you input a list of genes and select the Use reference without annotations.
For eukaryotes, it is more complex because the Workbench needs to know the intron-exon structure as explained in the introduction. This means that you need to have a reference genome with annotations of type mRNA and gene (you can see the annotations of a sequence by opening the annotation table).
You can obtain an annotated reference sequence in different ways:
- Download the sequences from NCBI from within the Workbench (see GenBank search). Figure 27.6 shows an example of a search for the human refseq chromosomes.
- Retrieve the annotated sequences in supported format, e.g. GenBank format, and Import (
 ) them into the Workbench.
) them into the Workbench.
- Download the unannotated sequences, (e.g. in fasta format) and annotate them using a GFF/GTF file containing gene and mRNA annotations (learn more at www.clcbio.com/clc-plugin/annotate-sequence-with-gff-file/). Please do not over-annotate a sequence that is already marked up with gene and mRNA annotations unless you are sure that the annotation sets are exclusive. Overlapping gene and mRNA annotations will lead to useless RNA-Seq results.
You need to make sure the annotations are the right type. GTF files from Ensembl are fully compatible with the RNA-Seq functionality of the CLC Genomics Workbench: ftp://ftp.ensembl.org/pub/current_gtf/. Note that GTF files from UCSC cannot be used for RNA-Seq since they do not have information to relate different transcript variants of the same gene.
If you annotate your own files, please ensure that you use annotation types
geneand, if it is a eurkarote,mRNA. To annotate with these types, they must be spelled correctly, and the RNA part of mRNA must be in capitals. Please see see section 10.3annotation table.
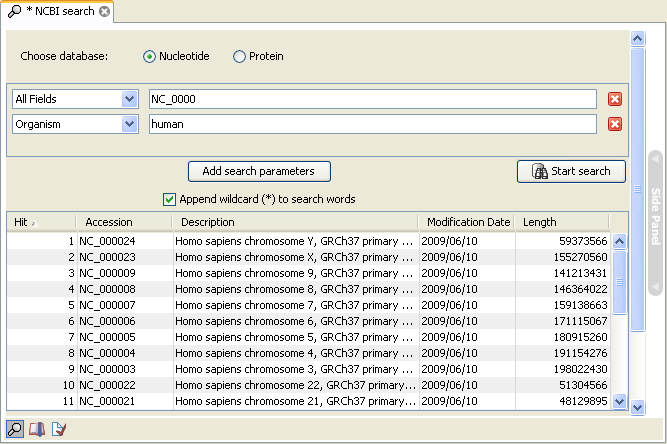
Figure 27.6: Downloading the human genome from refseq.