Definition of RPKM
RPKM, Reads Per Kilobase of exon model per Million mapped reads, is defined in this way [
Mortazavi et al., 2008]:
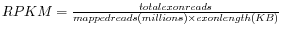
.
- Total exon reads
- This is the number in the column with header Total exon reads in the row for the gene. This is the number of reads that have been mapped to a region in which an exon is annotated for the gene or across the boundaries of two exons or an intron and an exon for an annotated transcript of the gene. For eukaryotes, exons and their internal relationships are defined by annotations of type mRNA.
- Exon length
- This is the number in the column with the header Exon length in the row for the gene, divided by 1000. This is calculated as the sum of the lengths of all exons annotated for the gene. Each exon is included only once in this sum, even if it is present in more annotated transcripts for the gene. Partly overlapping exons will count with their full length, even though they share the same region.
- Mapped reads
- The sum of all the numbers in the column with header Total gene reads. The Total gene reads for a gene is the total number of reads that after mapping have been mapped to the region of the gene. Thus this includes all the reads uniquely mapped to the region of the gene as well as those of the reads which match in more places (below the limit set in the dialog in figure 27.4) that have been allocated to this gene's region. A gene's region is that comprised of the flanking regions (if it was specified in figure 27.4), the exons, the introns and across exon-exon boundaries of all transcripts annotated for the gene. Thus, the sum of the total gene reads numbers is the number of mapped reads for the sample. This number can be found in the RNA-seq report's table 3.1, in the 'Total' entry of the row 'Counted fragments'. (The term 'fragment' is used in place of the term 'read', because if you analyze paired reads and have chosen the 'Default counting scheme' it is 'fragments' that is counted, rather than reads (two reads in a pair will be counted as one fragment).