QC, Assemble and Bin Pangenomes
The QC, Assemble and Bin Pangenomes template workflow guides you through the key steps to analyze whole-genome shotgun metagenomic reads and assign them to clusters of sequences (bins) using the tools Bin Pangenomes by Taxonomy and Bin Pangenomes by Sequence. The inputs to the workflow are short reads belonging to a single metagenome sample (can be split in multiple sequence lists).
To run the workflow, go to:
Workflows | Template Workflows (![]() ) | Microbial Workflows (
) | Microbial Workflows (![]() ) | Metagenomics (
) | Metagenomics (![]() ) | Taxonomic Analysis (
) | Taxonomic Analysis (![]() ) | QC, Assemble and Bin Pangenomes (
) | QC, Assemble and Bin Pangenomes (![]() )
)
- Specify the sample you would like to analyze.
- Specify a Trim adapter list if your sequences contain adapters (see Adapter trimming).
- Specify the minimum contig length, the type of de novo assembly you wish to perform (fast, or optimized for longer contigs), and whether you wish to perform scaffolding (figure 2.5).
- For taxonomic binning of the assembled contigs, a Taxonomic Profiling Index must be provided (figure 2.6). Reference databases can be obtained by using the Download Curated Microbial Reference Database tool (Download Curated Microbial Reference Database) or Download Custom Microbial Reference Database tool (Download Custom Microbial Reference Database). For custom reference databases, indexes can be built with the Create Taxonomic Profiling Index tool (Create Taxonomic Profiling Index).
- In the next dialog (figure 2.7), configure the parameters for the Bin Pangenomes by Sequence tool. You can set the minimum contig length to exclude shorter contigs, as binning by sequence requires longer sequences for good results. You can also choose the maximum number of iterations that should be performed, and how to label singletons (bins with a single contig).
- In the "Create Sample Report" step various summary items have been set. These are guidelines to help evaluate the quality of the results (see Create Sample Report).
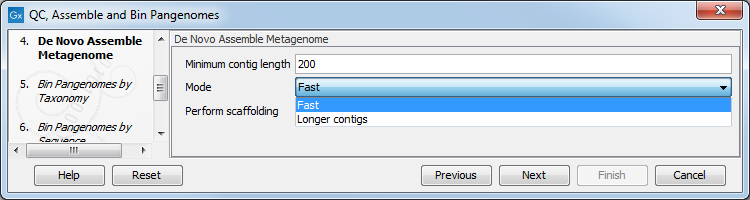
Figure 2.5: Parameters for the De Novo Assemble Metagenome tool.
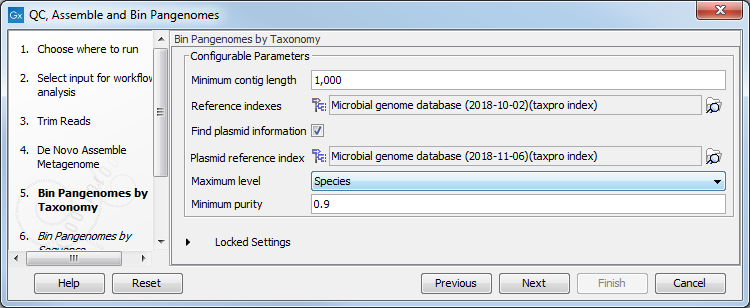
Figure 2.6: Select the reference index for Bin Pangenomes by Taxonomy.
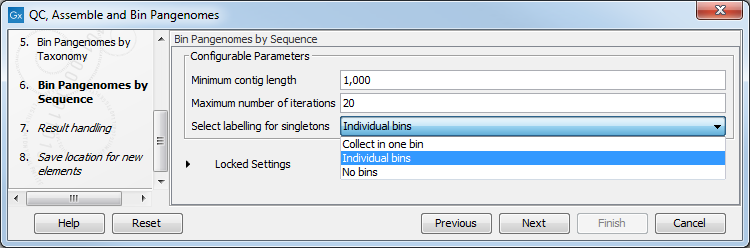
Figure 2.7: Configure the Bin Pangenomes by Sequence.
The workflow produces the following outputs:
- Reads binned by taxonomy. Folder containing sequence list(s) of reads binned by Bin Pangenomes by Taxonomy.
- Reads binned by sequence. Folder containing sequence list(s) of reads binned by Bin Pangenomes by Sequence.
- QC & Reports. Folder containing the individual reports generated during the analysis.
- Binned contigs. A combined sequence list of all the contigs binned by either taxonomy or sequence.
- Sample report. The sample report is curated to contain the most important information for analysis interpretation. All full reports are linked throughout the Sample report or can be found in the QC & Reports folder. The Sample report icon will be colored based on whether Summary item thresholds were met. See the "Quality control" section in the sample report for specifics.
The Sample report should be inspected in order to determine whether the quality of the sequencing reads and the analysis results are acceptable.
Additionally, you will find the "De novo assemble metagenome report" in the "QC & Reports" subfolder. For a detailed description, see (De Novo Assemble Metagenome output).
Individual bins can be extracted from the sequence and contig lists by filtering by the bin label in the "Assembly_ID" column, either manually in the table view of the sequence list or by using Filter on Custom Criteria or Split Sequence List. Contigs can be used for downstream analysis such as reference-based assembly (or re-assembly), functional analysis, typing etc.