Identify Viral Integration Sites
The Identify Viral Integration Sites tool searches for likely viral/host integration events. The tool works by searching for regions with reads with unaligned ends and/or discordant paired reads, where one read in the pair maps to the host, and the other read maps to a virus.
Notice: this tool can only be used for protocols such as hybrid capture, which specifically enriches for viral genomes while capturing at least some chimeric reads that map to both host and virus genomes.
The approach is the following:
- First, the input reads are mapped simultaneously against the host genome (e.g. human) and a viral database. Internally, the reads are mapped using the 'Find Best References using Read Mapping' tool. Any ambiguous reads are randomly assigned, corresponding to the standard "Non-specific match handling = Map randomly" read mapper option. This produces a host read mapping, and read mappings for all identified viruses. These read mappings are then scanned for potential breakpoints ends, which are the positions showing a pattern of unaligned ends.
- The potential breakpoint ends are filtered based on the following criteria:
- The number of reads with unaligned ends must be higher than the user-specified criteria
- The number of reads with unaligned ends must be more than 5% of the maximum for the position with the highest number of unaligned ends for the chromosome/virus.
- For the host, we collect and map all the unaligned ends for a given position against the viral genomes. Then we look at the position where the majority of the reads map on the viral genome, and check if there is a potential breakpoint within 50 bp of that position. Notice: we choose the closest viral breakpoint, and we always choose the read mapping position where the majority of reads map.
- Finally, we look at the broken read pairs on the host genome, where one read was within 500 bp of the host breakpoint (and on the same side as the aligned part of the reads found during the scan for unaligned ends), while the other read in the pair mapped to the virus. If this number of broken reads is larger than a user-specified threshold, the host/virus breakpoint ends are considered a sound match, and we add the host/virus breakpoint to our list of identified breakpoints.
To launch the Identify Viral Integration Sites tool, go to:
Tools | Microbial Genomics Module (![]() ) | Metagenomics (
) | Metagenomics (![]() ) | Taxonomic Analysis (
) | Taxonomic Analysis (![]() ) | Identify Viral Integration Sites (
) | Identify Viral Integration Sites (![]() )
)
One or more single or paired-end read files can be provided as input.
After selecting the input reads, it is possible to specify the host and virus references, and adjust the detection parameters, see (figure 6.6).
The following parameters are available:
- Viral references The viral sequences. The breakpoints identified from the read mappings against the human reference will be tested against these sequences.
- Viral annotations Annotations, such as a Gene or CDS track for the viral sequences. Notice, that these annotations can also be present on the viral sequence input if this is a sequence list. In this case, specifying the annotations here will be used instead of any annotations present on the viral sequence list.
- Host references The host sequences.
- Host annotations Annotations, such as a Gene or CDS track for the host sequences. Notice, that these annotations can also be present on the host sequence input if this is a sequence list. In that case, specifying the annotations here will overwrite any annotations present on a host sequence list.
- Minimum number of reads on a virus At least this many reads must map to a virus before it is included in the analysis.
- Minimum relative virus abundance to most abundant virus A reference must have at least this fraction of the reads of the most abundant virus.
- Minimum virus coverage The minimum number of nucleotides mapped to the virus reference divided by the reference length before it is included in the analysis.
- Minimum reads with unaligned ends supporting site The minimum number of reads required with an unaligned end starting at the same position.
- Minimum host/virus broken pairs supporting site The minimum number of paired reads spanning the breakpoint site, where one read maps to the virus, and the other to the host.
- Minimum ratio between unaligned and aligned The minimum ratio between reads supporting a breakpoint, and reads with no unaligned ends. This is only checked for the host genome.
- Minimum unaligned end length Minimum length of unaligned ends to be considered as supporting a breakpoint.
- Nearby genes distance If host genes are located within this distance of an integration event (in basepairs) they are reported in the table view, and in the report.
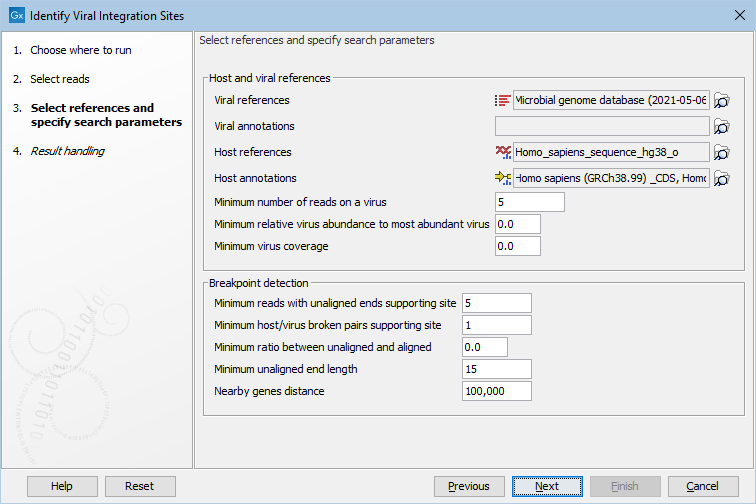
Figure 6.6: Select references and adjust detection options.
The final step is to specify the output objects, see (figure 6.7). The following options are available:
- Create breakpoint visualization Creates a graphical visualization and a table with breakpoints. This element is explained in more detail in the next section.
- Create report Creates a summary report.
- Create host breakpoint tracks Creates a feature track with detected breakpoints.
- Create viral breakpoint tracks Creates a feature track with detected breakpoints for the identified viruses.
- Create host mappings Creates a read mapping for the host references.
- Create viral mapping tracks Creates a read mapping for the (detected) viral references.
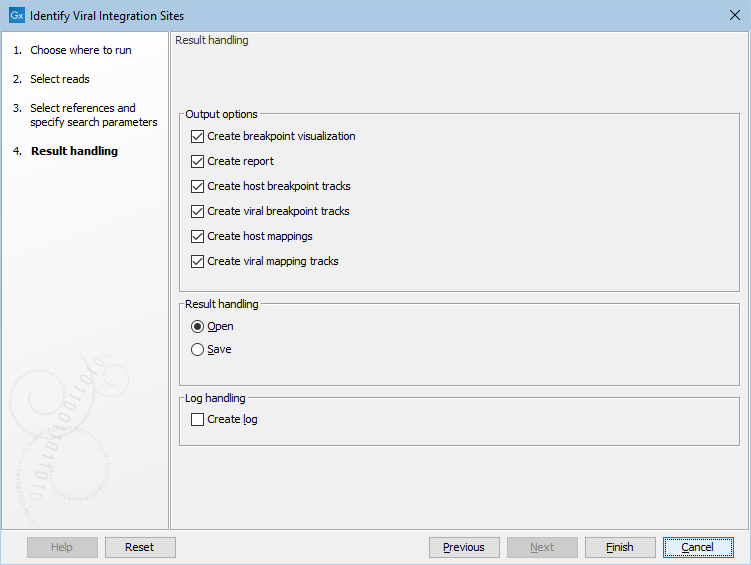
Figure 6.7: Select output options.
Subsections