Bin Pangenomes by Sequence
Binning by sequence is done irrespective of a database, only depending on content and coverage. To have both sources of information available, the Bin Pangenomes by Sequence tool takes read mappings to contigs as input, where there should be one read mapping per technical replicate (each mapping to the same contigs) in order to make most use of coverage information across all samples. However, if read mappings are not available, the Bin Pangenomes by Sequence tool also takes plain sequence lists of contigs as input.
The Bin Pangenomes by Sequence algorithm is based on the MetaBAT[Kang et al., 2015] and SCIMM[Kelley and Salzberg, 2010] algorithms with several modifications:
- A logistic regression model (simliar to MetaBAT) may use a variable number of parameters. The number of trusted parameters is adjusted to the number of contigs in a bin and the parameters are adjusted during the algorithm. Only Kmer features are used.
- The interpolated Markov Models of SCIMM are replaced by variable order markov models.
- Random Projections are used to speed up the search for the centers in the proximity of a contig in combined Kmer-Coverage space.
- Poisson-Mixture models are used to fit the coverage distribution on contigs.
The tool was designed for sample sizes on the order of 100 000 contigs. It does not support substantially larger data sets.
To start the tool, go to:
Tools | Microbial Genomics Module (![]() ) | Metagenomics (
) | Metagenomics (![]() ) | Taxonomic Analysis (
) | Taxonomic Analysis (![]() ) | Bin Pangenomes by Sequence (
) | Bin Pangenomes by Sequence (![]() )
)
The Bin Pangenomes by Sequence takes one sequence list of contigs or one read mapping per sample as input (figure 6.4).
Figure 6.4: Select the contigs or read mappings.
In the next dialog (figure 6.5), the several parameters can be specified:
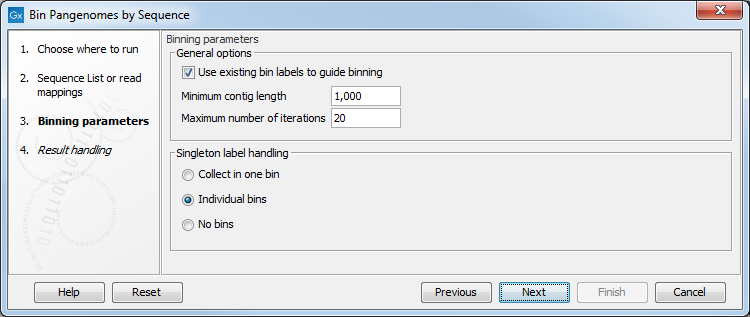
Figure 6.5: Configuration of the Bin Pangenomes by Sequence.
- Use existing labels to guide binning. May be used to improve binning quality and speed. For read mapping inputs, labels assigned to the reads by the Bin Pangenomes by Taxonomy are used, while for sequence list inputs the "Assembly ID" attribute on the contigs is used.
- Minimum contig length. Specifies the minimal length for contigs to be considered (should be at least 1000 to obtain decent bin qualities
- Maximum number of iterations. Specifies how many purification steps at most should be made.
- Singleton label handling. Decides whether singletons should be collected in one bin, kept in individuals bin, or not included in any bins.
The tool outputs a contig list with the assigned bin of each sequence listed in the Assembly_ID column. Additionally, the following outputs can be selected in the "Result handling" dialog:
- Create annotated sequence list (only if the input was read mappings). Outputs one sequence list per read mapping input, with each read assigned to the bin of the contig it belongs to.
- Output best bins separately. Outputs a contig list per bin for the number set in "Number of bins" below. Best bins are determined as those having more nucleotides.
- Create report. Outputs the sequence binning report.