Data QC and Remove Background Reads
The Data QC and Remove Background Reads template workflow performs trimming of reads, creates a QC report and cleans the dataset from background DNA, leaving back only the reads that match the reference genome(s).
To run the workflow, go to:
Workflows | Template Workflows (![]() ) | Microbial Workflows (
) | Microbial Workflows (![]() ) | Metagenomics (
) | Metagenomics (![]() ) | Taxonomic Analysis (
) | Taxonomic Analysis (![]() ) | Data QC and Remove Background Reads (
) | Data QC and Remove Background Reads (![]() )
)
- Specify the sample(s) or folder(s) of samples you would like to analyze.
- Specify a Trim adapter list if your sequences contain adapters (see Adapter trimming).
- In the "Taxonomic Profiling" step (figure 2.1), select the "Species of interest taxpro index" you will use to map the reads, and the "Background taxpro index". Reference databases can be obtained by using the Download Curated Microbial Reference Database tool (Download Curated Microbial Reference Database) or Download Custom Microbial Reference Database tool (Download Custom Microbial Reference Database). For custom reference databases, indexes can be built with the Create Taxonomic Profiling Index tool (Create Taxonomic Profiling Index).
- In the "Create Sample Report" step various summary items have been set. These are guidelines to help evaluate the quality of the results (see Create Sample Report).
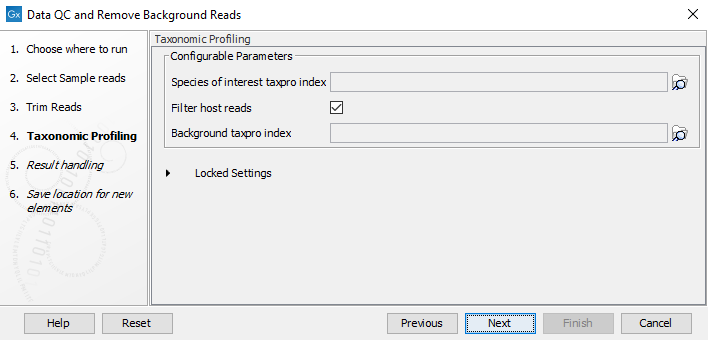
Figure 2.1: Select the species of interest taxpro index and a background taxpro index to remove possible contamination.
The workflow produces the following outputs:
- Cleaned reads. Folder containing trimmed reads mapping to the species of interest taxpro index of reference genome(s).
- Background reads. Folder containing reads mapping to the background taxpro index.
- Unmapped reads. Folder containing reads not mapping to the species of interest or the background taxpro index.
- QC & Reports. Folder containing the individual reports generated during the analysis.
- Sample report. The sample report is curated to contain the most important information for analysis interpretation. All full reports are linked throughout the Sample report or can be found in the QC & Reports folder. The Sample report icon will be colored based on whether Summary item thresholds were met. See the "Quality control" section in the sample report for specifics.
The Sample report should be inspected in order to determine whether the quality of the sequencing reads and the analysis results are acceptable.