QIAseq 16S/ITS Demultiplexer
The Panel Support section offers a tool to demultiplex NGS reads of different bacterial variable and fungal ITS regions obtained with the QIAGEN QIAseq 16S/ITS Screening and Region panels. Using this tool, sequences are associated with a particular region when they contain a match to a particular barcode. Sequences that do not contain a match to any of the barcode sequences provided are classified as not grouped.To run the tool, go to:
Microbial Genomics Module (![]() ) | Panel Support (
) | Panel Support (![]() ) | QIAseq 16S/ITS Demultiplexer (
) | QIAseq 16S/ITS Demultiplexer (![]() )
)
In the first dialog, window select the reads you wish to demultiplex and click Next. It is possible to run the tool in batch mode.
In the second dialog, select the barcodes used to demultiplex the sequences, from a predefined list (see the list in figure 19.1) or from a custom list. Select only barcodes corresponding to the primers used for library construction.
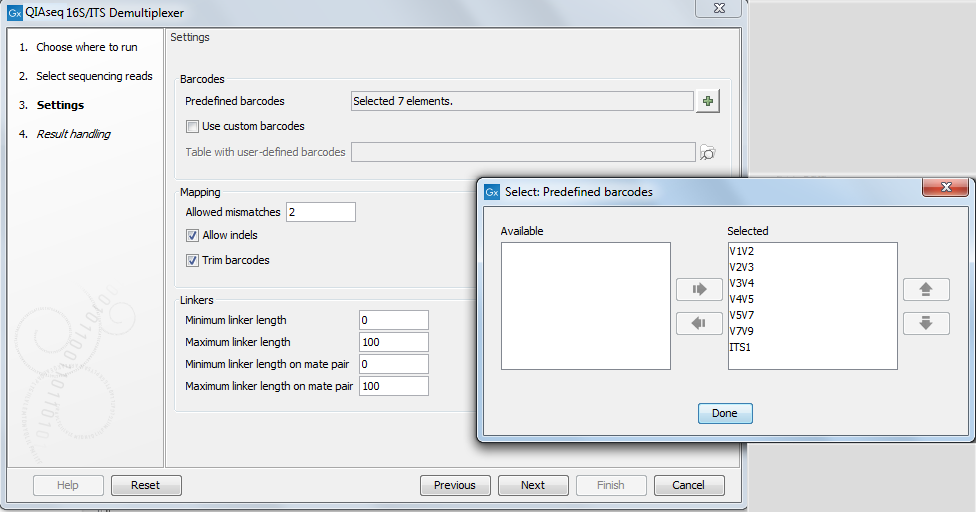
Figure 19.1: Set the parameters to demultiplex.
If you choose to use a table of custom barcodes (figure 19.2), you need to specify an Excel or a CSV file previously saved in the Navigation Area. The table will be different when setting barcodes for single or paired reads: for single reads, the first column defines the barcode name, the second contains the barcode sequence. For paired reads, an additional third column contains the reverse complement of the barcode sequence.
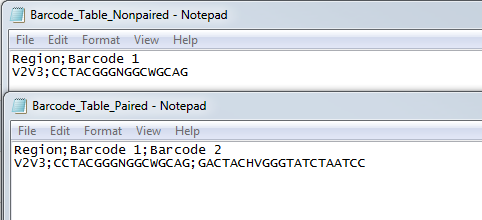
Figure 19.2: Examples of CSV custom barcodes files for paired and single reads.
The following parameters for demultiplexing are also available in this dialog:
- Mapping
- Allow mismatches: decide how many mismatches are allowed between the sequence and the barcode
- Allow indels
- Trim barcodes
- Linkers: also known as adapters, linkers are sequences which should just be ignored - it is neither the barcode nor the sequence of interest. For this element, you simply define its length.
- Minimum linker length
- Maximum linker length
- Minimum linker length on mate pair
- Maximum linker length on mate pair
In the Result handling window, you can choose to Create a report (see figure 19.3), and Save a sequence list of all ungrouped sequences.
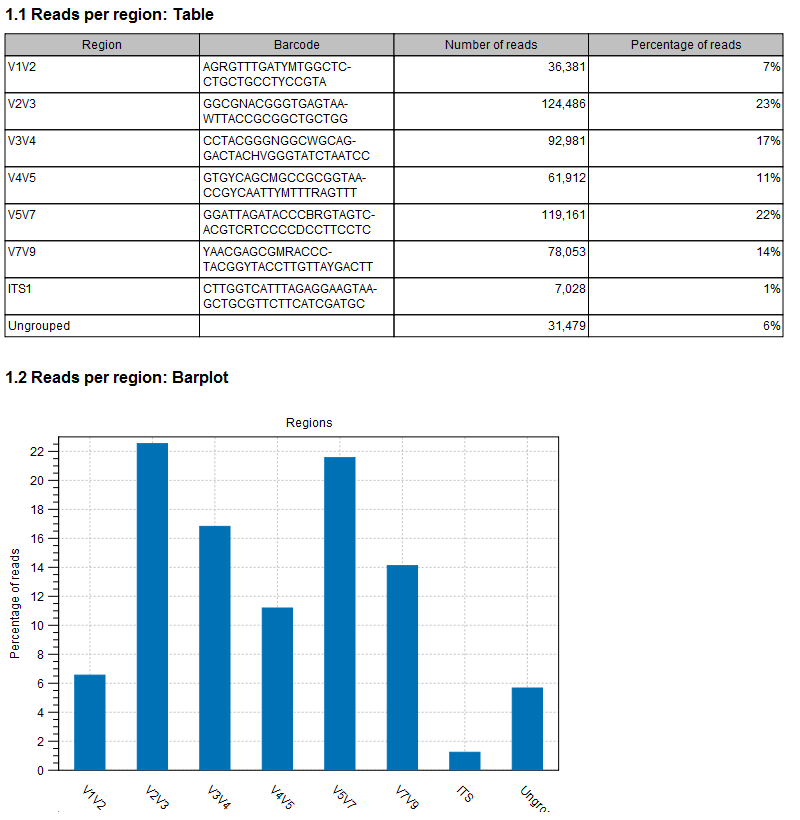
Figure 19.3: An example of demultiplexing report.
The main output are sequence lists for each different regions/barcodes. These sequence lists can be used as input for the Data QC and OTU Clustering workflow, that will generate an output table displaying the OTUs abundances for each region. Note that the Trim Reads step of the workflow will automatically detect and trim the remaining read-through barcodes found on paired-end reads and not discarded by the demultiplexer. However, if you are working with single reads, mate-paired reads or data of low quality, it is recommended to specify a trim adapter list containing all barcodes in the Trim Reads step of the workflow.