Type With MLST Scheme
The Type With MLST Scheme tool is used for assigning a sequence type to an isolate.
Type With MLST Scheme is available from:
Tools | Microbial Genomics Module (![]() ) | Typing and Epidemiology (
) | Typing and Epidemiology (![]() ) | MLST Typing (
) | MLST Typing (![]() ) | Type With MLST Scheme (
) | Type With MLST Scheme (![]() )
)
The tool takes a sequence list as input and will work with either raw NGS reads or an assembled genome. Note that if the input is raw NGS reads, and the tool reports multiple ambiguous sequence types, performing a standard De Novo Assembly might help to reduce noise and provide a more conclusive typing result.
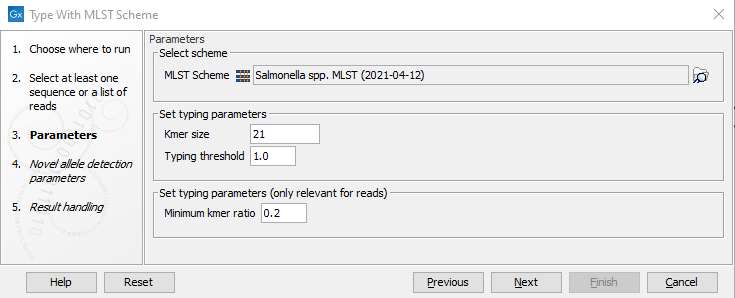
Figure 10.12: Specifying scheme and typing parameters.
In the next dialog step (figure 10.12), specify the scheme and the typing parameters. MLST schemes are available from Download MLST Scheme (see Download MLST Scheme).
Some MLST schemes contain sequence types with ambiguous bases. The Type With MLST Scheme tool does not support ambiguous bases and such sequence types will effectively be ignored.
The tool works by comparing the kmers in the input to the kmers in the alleles for the different loci.
The Kmer size determines the number of nucleotides in the kmer - raising this setting might increase specificity at the cost of some sensitivity.
The Typing threshold determines how many of the kmers in a sequence type that needs to be identified before a typing is considered conclusive. The default setting of 1.0 means that all kmers in all alleles must be matched. Lowering the setting to 0.99 would mean that on average 99% of the kmers in all the alleles of a given sequence type must be detected before the sequence type is considered conclusive. The typing threshold must be 0.5 or above.
When working with reads, the Type With MLST Scheme tool works by classifying allele calls as high-confidence and low-confidence calls to remove alternative allele calls for the same locus. The Minimum kmer ratio threshold gives the possibility to tweak the balance between high-confidence and low-confidence allele calls, e.g. decreasing this number will result in more high-confidence allele calls and thus more ambiguity in how an ST is assigned to the sample, conversely increasing this number will result in fewer high-confidence calls and may lead to no allele being called for a particular locus, which can make sequence type assignments less confident. Specifically, the kmer ratio is calculated as the number of observations for the least occurring kmer in an allele divided by the average number of observations for all kmers.
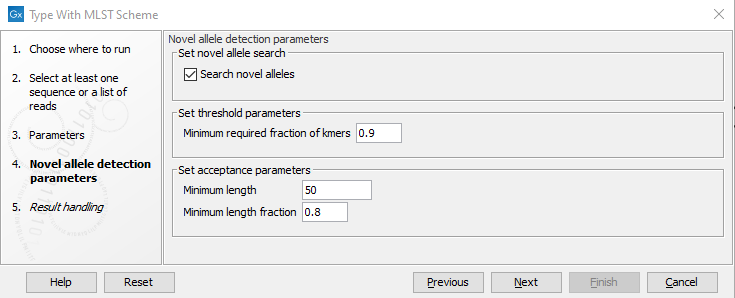
Figure 10.13: Specifying novel allele detection parameters.
The next step in the dialog determines how to handle novel alleles (figure 10.13): if the input isolate has loci with alleles that are not part of the scheme, it is possible to still detect the novel alleles. The novel alleles and the resulting new sequence type can then be added to the scheme using the Add Typing Results to MLST Scheme tool.
Novel alleles are detected as close hits to existing alleles in a locus. The Minimum required fraction of kmers determines how close a match must be: the default setting of 0.9 means that at least 90% of the kmers for an allele in a locus must be identified before the novel allele detection is initiated.
If the input to the tool is raw NGS reads, the tool will assemble the reads containing the kmers for the possible novel allele. If the input is already an assembled genome, the existing alleles for a locus will be mapped to the assembly to extract a novel allele.
After a candidate novel allele has been identified, it is aligned to the other alleles in the locus.
If the scheme has been built with the Check codon positions option of the Create MLST Scheme tool enabled (see Create MLST Scheme)), or if the scheme was imported with a specified genetic code (see Download MLST Scheme), the start and stop codons in the novel allele sequence are then identified, and the sequence is then trimmed to the start and stop codons that most closely match the length of the existing alleles in the locus. Alleles that contain both a start and a stop codon at the beginning and end, respectively, and pass the acceptance parameters (see below) will be marked as Complete in the output table from the tool.
The acceptance parameters describe the final consistency check: the novel allele must not contain a stop codon, it must be at least the Minimum length in nucleotides and have at least a length of the specified Minimum length fraction of the shortest allele in the locus before it is accepted.
Subsections