Classify Long Read Amplicons parameters
To run the Classify Long Read Amplicons tool, go to:
Tools | Microbial Genomics Module (![]() ) | Metagenomics (
) | Metagenomics (![]() ) | Amplicon-Based Analysis (
) | Amplicon-Based Analysis (![]() ) | Classify Long Read Amplicons (
) | Classify Long Read Amplicons (![]() )
)
Classify Long Read Amplicons takes single-end long-read amplicon reads as input. The data should be trimmed for adapters, barcodes, and preferably also primers. Quality scores are not needed for the tool to work and you do not need to quality trim the reads. Since the algorithm infers an error model based on the read alignments, samples from different runs should not be analyzed simultaneously. Instead, you can run samples in "Batch" mode.
In the wizard, three categories of parameter settings need to be set (figure 5.21):
- Select reference. Specify the reference database to be used. Reference databases can be downloaded with the Download Amplicon-Based Reference Database tool (Download Amplicon-Based Reference Database) or created by adding taxonomies to sequence lists using the Update Sequence Attributes tool (https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Update_Sequence_Attributes.html).
Reads receive the taxonomy available on the reference sequence assigned i.e., if the reference has any missing taxonomy levels, these will also be missing from the final abundance table. - Read alignment. Select whether to let the mapping algorithm set mapping parameters automatically based on the read platform used, or whether to manually override them. If choosing "Manual", the parameters can be set in the boxes below.
- Abundance estimation. Set the "Minimum relative abundance". Any taxa that have relative abundance below this threshold after the expectation maximization loop converges, will be removed and the abundance from these taxa will be reassigned to other probable taxa among the retained taxa.
Note: If you are going to perform differential abundance analysis (section 7.7) following the classification of amplicons it may be an idea to set "Minimum abundance threshold" to 0. Differential abundance analysis on few samples with few features (taxa) can cause poor dispersion estimates. Should you later want to remove low abundance taxa, you can create an abundance subtable after filtering the table manually using the advanced filters in the top right of the table view.
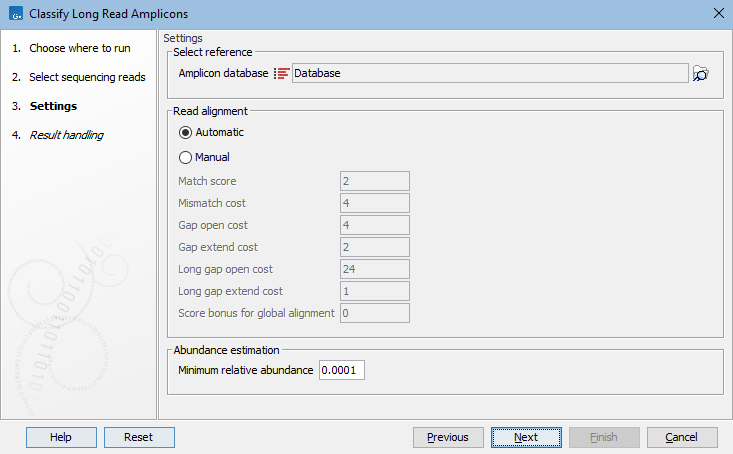
Figure 5.21: Classify Long Read Amplicons parameter settings.