Detect Amplicon Sequence Variants parameters
To run the Detect Amplicon Sequence Variants tool, go to
Tools | Microbial Genomics Module (![]() ) | Metagenomics (
) | Metagenomics (![]() ) | Amplicon-Based Analysis (
) | Amplicon-Based Analysis (![]() ) | Detect Amplicon Sequence Variants (
) | Detect Amplicon Sequence Variants (![]() )
)
Select the single- or paired-end sequence lists to be analyzed. For paired reads, pairs should have a minimum overlap of 12 bases. Data should be trimmed beforehand to remove adapters and poor quality nucleotides. This can be done using Trim Reads (https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Trim_Reads.html).
Set the Trim and filter parameters (figure 5.13):
- First/Second read length: Reads are trimmed to the given length from the 3' end. Reads shorter than this will be discarded. For paired reads you may set different values for the first and second read in a pair.
What values to set will depend on your sequencing protocol and how reads are trimmed prior to being used as input for the Detect Amplicon Sequence Variants tool. We recommend that you have a look at the Trim report section Read length before / after trimming if you are unsure about what value to set.
You must use the same length setting for all samples that will be compared in downstream analysis.
- Maximum expected errors per read: The maximum number of expected errors allowed for a read. Reads with more expected errors will be discarded.
- Remove chimeras: If selected, reads identified as chimeras will be discarded.
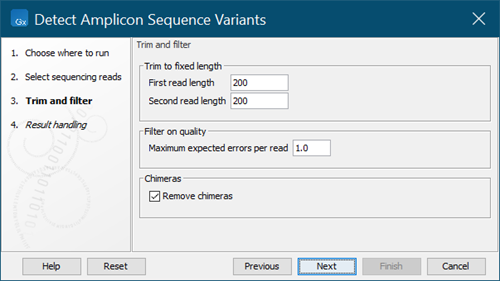
Figure 5.13: Trim and filter parameter settings