Detect Amplicon Sequence Variants and Assign Taxonomies
The Detect Amplicon Sequence Variants and Assign Taxonomies workflow processes reads from amplicon sequencing to yield a merged multi-sample (if applicable) ASV (Amplicon Sequence Variant) abundance table, and subsequently assigns taxonomies to the ASVs (amplicon sequence variants).
We recommend making preliminary evaluations of the read lengths and qualities, to decide on parameter settings like read length. This can be done by running a single sample through the workflow, and taking a look at the resulting trim report section Read length before / after trimming.
Launching the workflow
The Detect Amplicon Sequence Variants and Assign Taxonomies template workflow is available at:
Workflows | Template Workflows (![]() ) | Microbial Workflows (
) | Microbial Workflows (![]() ) | Metagenomics (
) | Metagenomics (![]() ) | Amplicon-Based Analysis (
) | Amplicon-Based Analysis (![]() ) | Detect Amplicon Sequence Variants and Assign Taxonomies (
) | Detect Amplicon Sequence Variants and Assign Taxonomies (![]() )
)
Launch the workflow and step through the wizard.
- Select the sequence list(s) containing the reads to process and click on Next.
- Select a Taxonomic Profiling Index and click on Next.
- The 'Configure batching' and 'Batch overview' steps can be left as is, or configured as described in Launching workflows individually and in batches.
- Select a Trim Adapter List if relevant for your application. The Trim Adapter List should correspond to the adapters used for sequencing. If no input is provided, the tool will skip the adapter trimming step. Click on Next.
- Choose the trim length to use for Detect Amplicon Sequence Variants and decide whether to remove chimeras by toggling the Remove chimeras box (figure 2.8). Click on Next.
The optimal read length setting will depend on the length of your reads after trimming. We recommend that you have a look at the trim report section Read length before / after trimming if you are unsure about what value to set.
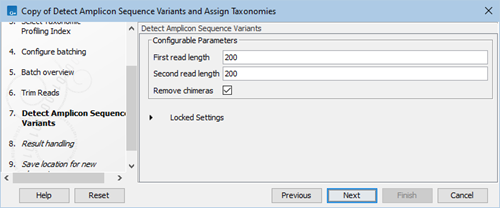
Figure 2.8: Wizard step for selecting read trim length and whether to remove chimeras in the Detect Amplicon Sequence Variants tool. - In the "Create Sample Report" step various summary items have been set. These are guidelines to help evaluate the quality of the results (see Create Sample Report).
- Various options and filters can be set for refining the merged abundance table (see Refine Abundance Table).
- Finally, select a location to save outputs to and click on Finish.
Workflow outputs
The batch-specific outputs provided by this workflow are:
- Sample report. The sample report is curated to contain the most important information for analysis interpretation. All full reports are linked throughout the Sample report or can be found in the QC & Reports folder. The Sample report icon will be colored based on whether Summary item thresholds were met. See the "Quality control" section in the sample report for specifics.
- Analysis results. Folder containing results output during analysis.
- ASV table. ASV abundance table with abundances for each detected ASV.
- ASV sequence list. Sequence list containing the detected ASVs.
- QC & Reports. Folder containing the individual reports generated during the analysis.
- All reports from the sample report are found here in their full length.
The combined outputs provided by this workflow are:
- Assign taxonomies report. Report output by Assign Taxonomies to Sequences in Abundance Table (Assign Taxonomies to Sequences in Abundance Table output). Is also in the Combined report.
- Refine abundance table report. Report output by Refine Abundance Table (Refine Abundance Table report). Is also in the Combined report.
- Combined report. Combined report of all sample reports and the Assign taxonomies and Refine abundance table reports. The combined report contains all quality control information and analysis results. The combined report icon will be colored based on whether Summary item thresholds were met in each sample. See the "Quality control" section in the combined report for specifics.
- Merged and refined abundance table with taxonomies. Abundance table containing abundance results with taxonomy from all samples input into the workflow. If parameters were set in the Refine Abundance Table step, the abundance table will have been refined accordingly.
The Combined report should be inspected in order to determine whether the quality of the sequencing reads and the analysis results are acceptable.