Find Best References using Read Mapping
Find Best References using Read Mapping takes as input one or more sequence lists (![]() ). The tool maps the input reads to a set of target reference sequences and outputs the best target references, i.e., the references for which the reads provide the strongest evidence.
). The tool maps the input reads to a set of target reference sequences and outputs the best target references, i.e., the references for which the reads provide the strongest evidence.
To run the tool, go to:
Tools | Microbial Genomics Module (![]() ) | Typing and Epidemiology (
) | Typing and Epidemiology (![]() ) | Find Best References using Read Mapping (
) | Find Best References using Read Mapping (![]() )
)
References
The following options can be configured in the References dialog (figure 8.6):
- Target references. Sequence list(s) (
 ) or sequence track(s) (
) or sequence track(s) ( ) containing the target reference sequences.
) containing the target reference sequences.
- Host references. Optional sequence list(s) () or sequence track(s) () containing host or background reference sequences. Reads that map better to host references are assigned to the host and excluded from the target references mapping. If host references are not provided, reads originating from the host or background may instead contribute support to similar target references.
- Reference handling. Specifies how target and host sequences are grouped and treated as references during the analysis.
- Treat each sequence as a reference. Each sequence is treated as a separate reference.
- Treat each assembly as a reference. Sequences sharing the same "Assembly ID" are grouped and treated as a single reference. Sequences with an empty "Assembly ID" are treated as separate references. Recommended when working with segmented references.
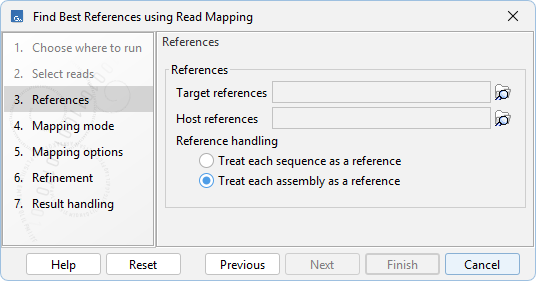
Figure 8.6: Options for configuring references.
Target and host references are checked for uniqueness:
- If multiple references have the same name and sequence, only one of them is used.
- If multiple references have the same name but different sequences, their names are updated to ensure uniqueness.
Mapping mode
The type of mapping to be performed can be configured in the Mapping mode dialog (figure 8.7):
- Standard mapping. Reads are mapped to all references in a single step. Reads mapping equally well to more than one reference are handled according to Non-specific match handling. See Non-specific matches for details.
Mapping statistics are calculated for candidate target references with at least one mapped read and are then used when filtering the references as configured in the Refinement dialog.
Recommended for routine analyses where speed is important and the target references are expected to be sufficiently distinct.
- High-sensitivity mapping. Reads are first mapped to all references, after which the mapping to the target references is refined using BLAST.
If host references are provided, reads mapping equally well to both host and target references are handled according to Non-specific match handling:
- Map to target. Reads are assigned to the target. Recommended for higher sensitivity.
- Map to host. Reads are assigned to the host. Recommended for higher specificity.
Reads mapping equally well to multiple target references, or to multiple host references, are assigned randomly.
Reads mapping to a target reference are then aligned against target references using BLAST. The following options can be configured for the BLAST refinement:
- Maximum coverage. Reads used for BLAST refinement are randomly downsampled so that no region has coverage exceeding this threshold.
- Minimum percent identity (%). Minimum percentage identity over the aligned region required for a read to be considered mapped.
- Minimum overlap (%). Minimum percentage of the read length that must be covered by the alignment for a read to be considered mapped.
See High-sensitivity mapping for details.
Mapping statistics are calculated from the BLAST mapping for candidate target references with at least one mapped read and are then used when filtering the references as configured in the Refinement dialog.
Recommended when reference sequences are similar or when low-abundance target references must be detected.
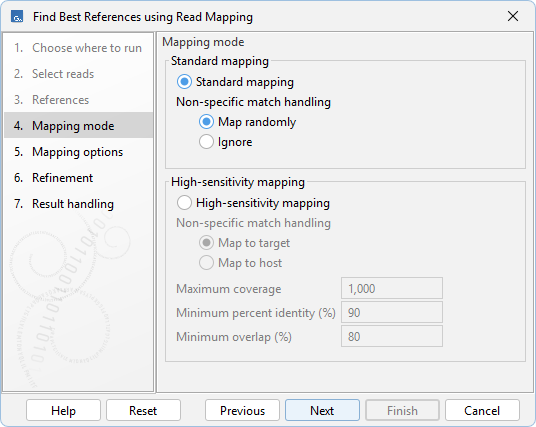
Figure 8.7: Options for configuring how mapping should be performed.
Mapping options
See Mapping options for details about the options in the Mapping options dialog.
Refinement
How the best target references are identified among candidate target references based on the calculated mapping statistics, and how their mapping is refined, can be configured in the Refinement dialog (figure 8.8):
- References refinement
Filtering is applied at the assembly level rather than to individual target references when using Treat each assembly as a reference.
- Filter by similarity. When checked, discard references with similarity above Maximum similarity to another reference with more mapped reads. Similarity is approximated from the shared canonical 31-mers of each target reference sequence. Recommended for increased coverage when target references are similar.
- Filter by read count. When checked, discard references with fewer mapped reads than Minimum read count.
- Filter by relative abundance. When checked, discard references whose abundance, measured as the number of mapped reads relative to the highest-ranked reference, is below Minimum relative abundance. References are ranked by the number of mapped reads.
- Filter by coverage fraction. When checked, discard references whose fraction of bases covered by at least one read is below Minimum coverage fraction.
- Filter by average coverage. When checked, discard references for which the number of mapped bases divided by the reference length is below Minimum average coverage.
- Filter by number of references. When checked, limit the number of best target references to Maximum number of references. References are ranked by the number of mapped reads.
- Mapping refinement
- Remap reads to best target references. When checked, reads that mapped to a target reference in the first mapping step are used in a final mapping to the best target references. This can improve both coverage fraction and average coverage for the best target references.
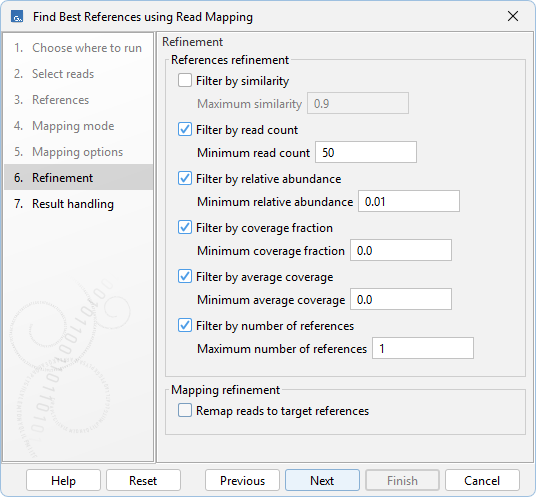
Figure 8.8: Options for configuring references and mapping refinement.
Subsections