Join Nearby Variants
The Join Nearby Variants tool merges variants that are more than one nucleotide apart and within three nucleotides of each other into larger microhaplotypes. Because the tool merges variants regardless of their zygosity and frequency, it is most suited for use on isolates from monoploid organisms, such as most viruses and bacteria.
Merging variants allows amino acid changes to be more precisely determined by the Amino Acid Changes tool.
To run the tool, go to
Tools | Microbial Genomics Module (![]() ) | Drug Resistance Analysis (
) | Drug Resistance Analysis (![]() ) | Join Nearby Variants (
) | Join Nearby Variants (![]() )
)
The tool takes a variant track as input.
The following parameters are available (figure 13.10):
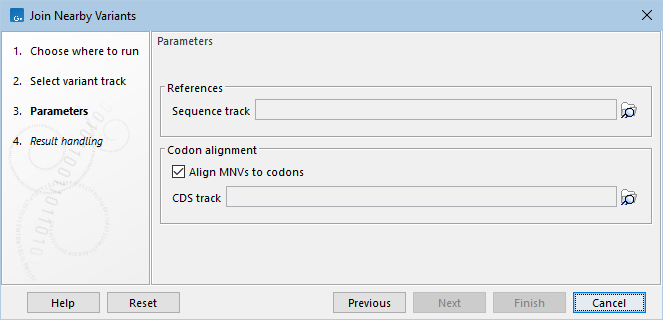
Figure 13.10: Join Nearby Variants parameter settings.
- Sequence track. The genome against which the variants were called. This is needed to fill in spaces between variants being called with reference nucleotides.
- Align MNVs to codons. When checked, MNVs are split into smaller MNVs and SNVs at codon boundaries. If a variant overlaps multiple CDS regions that have different reading frames, then the variant will be split in multiple ways: one for each reading frame.
- CDS track. The CDS track is required to determine codon boundaries when aligning MNVs to codons.
Note: The Codon alignment option is primarily intended for matching variants against variant databases from outside the CLC Genomics Workbench. Databases of known variants (such as the WHO drug resistance variant database, Reference Data Elements), can have variants given as amino acid substitutions i.e., given in triplets/codon MNVs as opposed to larger substitutions, like when called within the CLC Workbench. These longer variants must be split on codon boundaries in order to accurately match the database when using Filter against Known Variants or Annotate from Known Variants.
Detailed behavior
- Reference variants are removed from the output track.
- Overlapping variants are never merged with a nearby variant, because it is unclear which of the overlapping variants should be merged with the nearby variant.
- Variants that are adjacent are never merged. If adjacent variants were actually one longer variant, they would have been called as such by the variant detection tools (https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Variant_Detection_tools.html). Adjacent variants mean that the variants were not present on the same reads and thus should not be inferred to be a single variant.
- When variants are merged, the count, coverage etc. are the minimum of the observed values in the variants being merged. The frequency of the resulting variant is the minimum count divided by the minimum coverage. The zygosity of the resulting variant is unknown if any of the variants being merged have unknown zygosity, heterozygous if any of the variants being merged are heterozygous, and otherwise homozygous.
- The first variant in a group to be merged may be right-shifted to permit merging to occur. For example, if the reference is "ACGTACACACG" and the sample has "ACGTACACT", then there are three ways in which "AC" may be deleted. The tool right-shifts the deletion such that it is closest to the G -> T. The final variant called is then a replacement "ACG -> T".
- Variants will not be merged when they are on different sides of the origin of a circular chromosome, different sides of a splice junction, or either side of a ribosomal slippage (i.e. in a region where the CDS annotation overlaps itself by one or two nucleotides).