Annotate with BLAST
The Annotate with BLAST tool allows you to annotate a DNA sequence using a set of either protein reference sequences or nucleotide sequences. This tool can be used on sequences without any pre-existing annotations: it is not necessary to annotate the DNA sequences with genes or coding regions.
The tools can be used for various purposes, e.g. transferring annotations from a known reference, annotate the presence of AMR or virulence markers in a genome, or to filter contigs or sequences based on the presence of a set of genes.
If the reference sequences are protein sequences, the Annotate with DIAMOND tool may be used instead and is a faster option.
If the input sequences are already annotated with CDS annotations, it is also possible to use the Annotate CDS with Best BLAST Hit and Annotate CDS with Best DIAMOND Hit tools - see Annotate CDS with Best BLAST Hit for more information.
To start the analysis, go to:
Tools | Microbial Genomics Module (![]() ) | Functional Analysis (
) | Functional Analysis (![]() ) | Annotate with BLAST (
) | Annotate with BLAST (![]() )
)
The first wizard step (figure 12.2), specifies the reference and search parameters.
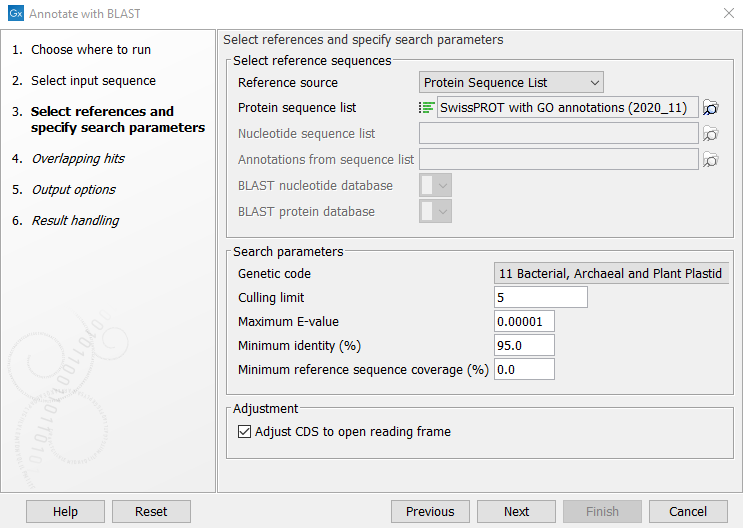
Figure 12.2: Selecting references and specifying search parameters
The following sources can be used to annotate the input sequences:
- Protein sequence list. The nucleotide input query will be searched against the sequences in the protein sequence list. The nucleotide input will be translated using the chosen genetic code. If the reference protein sequence list contains metadata, this metadata will be transferred to the resulting annotations on the input query sequence.
- Nucleotide sequence list. The nucleotide input query will be searched against the sequences in the nucleotide sequence list. If the reference nucleotide sequence list contains metadata, this metadata will be transferred to the resulting annotations on the input query sequence.
- CDS Annotations (blastx). This option uses a nucleotide sequence source with existing annotations as a source. All annotations are extracted, and translated to a protein database, which is searched similar to the Protein sequence list option. All qualifiers on the detected source annotations are transferred to the input query sequence.
- All Annotations (blastn). This option uses a nucleotide sequence source with existing annotations as a source. All annotations are extracted and searched similar to the Nucleotide sequence list option. All qualifiers on the detected source annotations are transferred to the input query sequence.
- BLAST nucleotide database. BLAST databases can be created using the Create BLAST database tool. This option works similar to the Nuclotide sequence list option, but can be faster, since the database can be reused. When using this option, the name and description of detected reference sequences are transferred to the input query sequence.
- BLAST protein database. BLAST databases can be created using the Create BLAST database tool, or downloaded using the Download BLAST database tool. This option works similar to the Protein sequence list option, but can be faster, since the database can be reused. When using this option, the name and description of detected reference sequences are transferred to the input query sequence.
As can be seen above, metadata (such as GO terms and taxonomy information) is handled differently depending on the database source:
- Protein / nucleotide sequence list. The sequence list may contain metadata, which can be inspected in the table view of the sequence list. Such metadata is transferred to the annotations created by this tool.
- CDS / all annotations. Annotations are transferred together with any metadata qualifiers the annotations contain.
- BLAST protein / nucleotide database. These database types are used for fast annotation with reference sequences and do not allow for metadata. If you require annotation with metadata, for instance when using an RNAcentral database with GO terms in order to build a functional profile, this option can not be used. Instead, the sequence list option must be used, even though it is slightly slower.
The search parameters can be modified using the following settings:
- Genetic code. The genetic code used when translating the nucleotide sequences before searching against the protein references.
- Culling limit. Hits that are contained within at least this number of higher-scoring hits will be excluded from the results. For very large databases (e.g., RNAcentral), it is recommended to keep this value low.
Note: This is an internal BLAST parameter applied before the Overlapping hits filtering settings.
- Maximum E-value. Maximum expectation value (E-value) threshold for accepting hits.
- Minimum identity (%). The minimum percent identity for a hit to be accepted. The percent identity is calculated based on the number of amino acid matches when using protein reference sequences (blastx), and based on the number of nucleotide matches when using nucleotide reference sequences (blastn). Notice. when annotating with a Protein sequence list of clustered sequences such as UniRef50, this should be lowered depending on the level of clustering in the database.
- Minimum reference sequence coverage (%). The minimum length fraction of the reference sequence that must be matched. Notice: this is length fraction per hit (HSP), and should be kept low when searching for non-contiguous matches.
Adjustment can be made to the annotation hits by the following setting:
- CDS adjustment. The found annotation hits will be adjusted to begin with a start codon, end with a stop codon and not contain any stop codons in between. The adjustment can extend the annotation to up to 110 percent of the length of the reference gene and will not be shorter than 90 percent of the reference gene length. The frame of the translation may change from the original alignment.
The next step (figure 12.3), determines how to handle when multiple overlapping hits are found on the input query sequence.
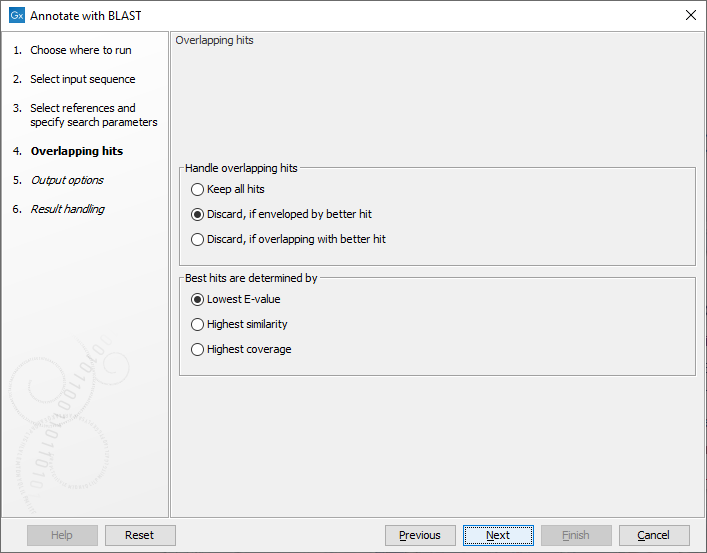
Figure 12.3: Settings for handling overlapping hits
The following options are available:
- Keep all hits. all hits that meet the search criteria are annotated on the input query sequence.
- Discard, if enveloped by better hit. If a hit covers the same region or part of the same region as a better hit, it is discarded.
- Discard, if overlapping with better hit. If a hit overlaps the same region as a better hit, it is discarded.
Best hits are determined by:
- Lowest E-value. hits with the lowest E-value are kept. Ties are resolved by highest similarity, subsequently highest coverage.
- Highest similarity. hits with the highest similarity are kept. Ties are resolved by lowest E-value, subsequently highest coverage.
- Highest coverage. hits with the highest coverage are kept. Ties are resolved by lowest E-value, subsequently highest similarity.
The output options step (figure 12.4), has the following options:
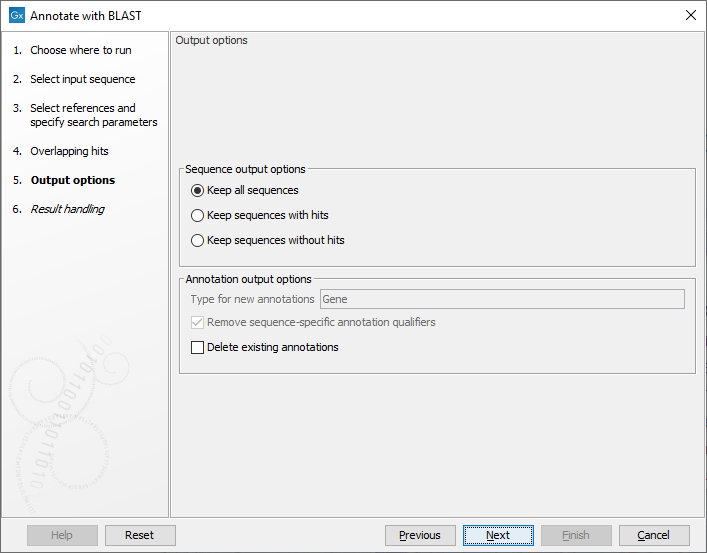
Figure 12.4: Specifying output options
- Type for new annotations. When using a protein database as source, all new annotations will be of type 'CDS'. However, when using a nucleotide sequence list, or a nucleotide sequence BLAST database, there is no general annotation type to apply. The default output annotation type will be 'Gene', but this can be customized if necessary.
- Remove sequence-specific annotation qualifiers. Annotation qualifiers such as 'translation' and 'codon_start' may no longer be accurate on the new annotations. This option removes such qualifiers.
- Delete existing annotations. Existing annotations on the input sequences will not be copied to the output sequences.
The following sequence output options are available:
- Keep all sequences
- Keep sequences with hits. This option can be useful for filtering input sequences for certain regions.
- Keep sequences without hits. This option can be useful for comparing sequence lists.
The final step controls which outputs are created. Notice, that reports can be aggregated using the Combine Reports tool.