Classify Whole Metagenome Data output
Click Next to select the outputs.
The tool outputs an abundance table. In addition, the following options are available:
- Collect reference database reads. Creates a sequence list for each input with reads that were assigned to the reference database.
- Collect host genome reads. Creates a sequence list for each input with reads that were assigned to the host genome.
- Collect unmapped reads. Creates a sequence list for each input with unassigned reads.
- Create report. Creates a summary report.
Sequence list output
The sequence list of reference database reads and host genome reads contain the Taxonomy attribute with the full taxonomy of the taxon to which the read was assigned.
Classify Whole Metagenome Data report
The Classify Whole Metagenome Data report (figure 6.14) contains information about the number of classified taxa per taxonomic level, and classification of reads.
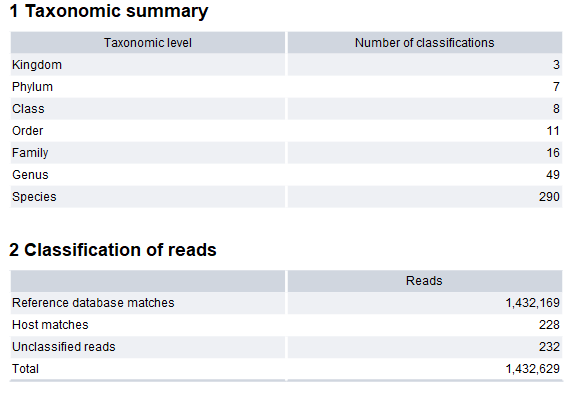
Figure 6.14: The Classify Whole Metagenome Data report.
- Taxonomic summary. The number of detected taxa for each taxonomic level.
- Classification of reads.
- Reference database matches. The number of reads assigned to the reference index, excluding host genome taxa.
- Host matches. The number of reads assigned to host genome taxa.
- Unclassified reads. The number of reads that could not be assigned to the reference index. If a large percentage of the reads are unclassified, it could mean that the sample is contaminated, or that the reference index is not comprehensive enough.