Find Resistance with PointFinder
Find Resistance with PointFinder identifies known antimicrobial resistance conferring mutations from reads. In contrast to the Find Resistance with Nucleotide Database tool that quantifies the occurrence of entire resistance conferring genes, the aim is to detect the presence of resistance conferring mutations in antimicrobial targets in both susceptible and resistant strains.
The presence of antimicrobial resistance conferring variants can be inferred by mapping reads to a point mutation database containing both wild type and known resistant mutants of antimicrobial target genes.
PointFinder databases can be downloaded using Download Resistance Database (Download Resistance Database).
To run Find Resistance with PointFinder, go to:
Tools | Microbial Genomics Module (![]() ) | Drug Resistance Analysis (
) | Drug Resistance Analysis (![]() ) | Find Resistance with PointFinder (
) | Find Resistance with PointFinder (![]() )
)
The tool accepts a nucleotide sequence or sequence list as input, followed by the selection of the point mutation database for the relevant pathogen.
For the read mapping step, several parameters are available (figure 13.1):
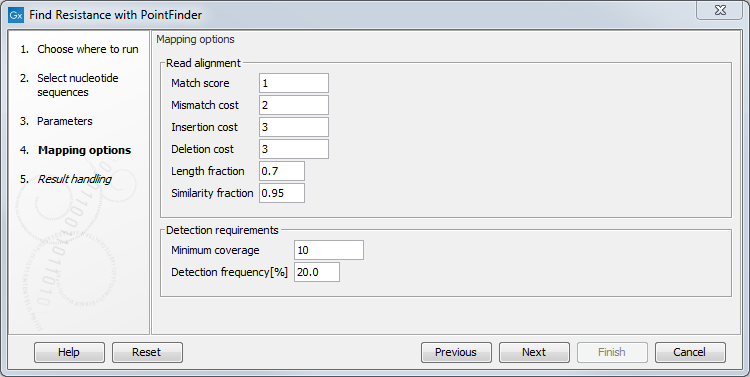
- Match score: score for a match
- Mismatch cost: cost for a mismatch
- Insertion cost: cost for an insertion
- Deletion cost: cost for accepting a deletion
- Length fraction: minimum length of the mapped portion of the read
- Similarity fraction: minimal fraction of matches in the mapped region
The tool first maps the reads to the specified database sequences. Next, the tool analyses the read mappings. For each reference sequence containing the variant, the tool verifies if the reads mapping to that reference or related references (e.g. references with an additional mutation) contain that variant. The result table will only list those entries from the database that have a minimum coverage and exceed the minimum frequency threshold. The tool also adds information:
- Minimum coverage: the number of reads covering the variant region. The coverage must be complete, e.g. all 3 nucleotides that constitute an amino acid change have to be covered.
- Detection frequency: The minimum allele frequency that is required to annotate a variant as being present in the sample.
The tool outputs a table containing information about the variants detected in the reads. The columns available in that table can be seen in figure 13.2 (which also shows an example of report output by the tool):
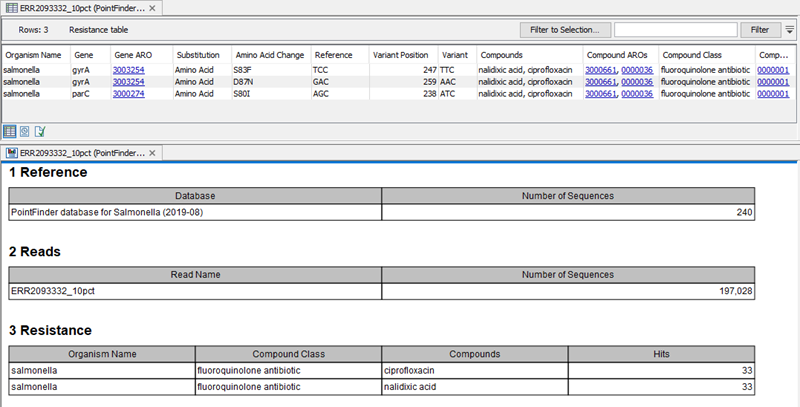
Figure 13.2: Table generated by Find Resistance with PointFinder, shown here together with the corresponding report.
By enabling the "Output annotated reads" option, the user can obtain a copy of the subset of reads that map to target variants. The reads will be annotated with the following annotations:
- Target: The name of the target reference (from the PointFinder database) where the read mapped to, e.g. "salmonella_gyrA_AAS_S_83_Y_TCC_247_TAC".
- Compound Class: The compound class to which the variant gives resistance, e.g. fluoroquinolone antibiotic.
- Compounds: The compound(s) class to which the variant gives resistance, e.g. colistin.
The resulting report contains information about the reads and the database that was used.