Compare MLST Typing Results
The Compare MLST Typing Results tool evaluates the genetic relatedness among multiple samples based on their MLST profiles. When working with several isolates, understanding how closely related these samples are can provide valuable insights into population structure and potential epidemiological links. This tool generates a distance matrix and graphical representations of similarities, allowing users to quickly identify clusters and patterns.
The Compare MLST Typing Results tool is available from:
Tools | Microbial Genomics Module (![]() ) | Typing and Epidemiology (
) | Typing and Epidemiology (![]() ) | MLST Typing (
) | MLST Typing (![]() ) | Compare MLST Typing Results (
) | Compare MLST Typing Results (![]() )
)
The tool takes one or more MLST Typing Results as input. Each result contains allelic profiles for a sample derived from a specific MLST Scheme. Pairwise distances are calculated between all samples based on the number of allele differences, the allelic distance. If the MLST Scheme includes sequence types (STs), the tool also searches for sequence types that have a low allelic distance to one or more input samples. This feature helps identify closely related STs that may not be present in the input dataset but are relevant for interpretation.
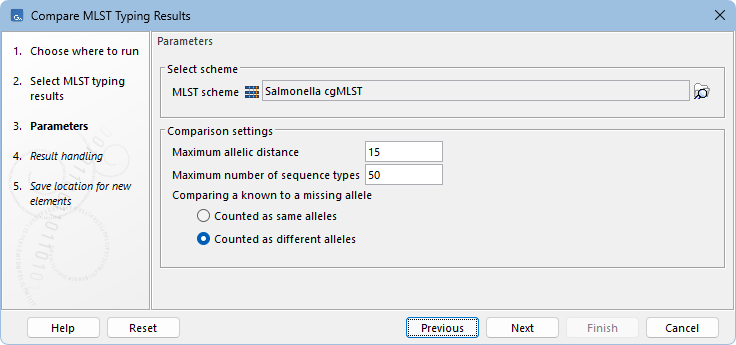
Figure 10.17: Compare MLST Typing Results parameters.
The tool has the following parameters (figure 10.17):
- MLST scheme. Select the same MLST Scheme used for typing the samples. Note that it is not possible to mix results for which different schemes were used.
- Maximum allelic distance. Sets the upper limit for the allelic distance relative to the input, for sequence types to be included in the comparison. Note that the sample inputs are always included, regardless of their distance to each other.
- Maximum number of sequence types. Sets the upper limit of sequence types to include in the comparison. For multiple inputs, this limit applies to the combined set of sequence types most similar to any of the provided samples, rather than per input.
- Comparing a known to a missing allele. Defines how missing alleles affect comparison. Counted as same alleles ignores missing loci in distance calculation, while Counted as different alleles treats them as mismatches, increasing allelic distance.
Subsections