Align OTUs with MUSCLE
To estimate Alpha and Beta diversity, OTUs must initially be aligned with the MUSCLE tool of the CLC Microbial Genomics Module:
Tools | Microbial Genomics Module (![]() ) | Metagenomics (
) | Metagenomics (![]() ) | Amplicon-Based Analysis (
) | Amplicon-Based Analysis (![]() ) | Align OTUs using MUSCLE (
) | Align OTUs using MUSCLE (![]() )
)
Choose an OTU abundance table as input. The next wizard window allows you to set up the alignment parameters with MUSCLE (figure 5.12).
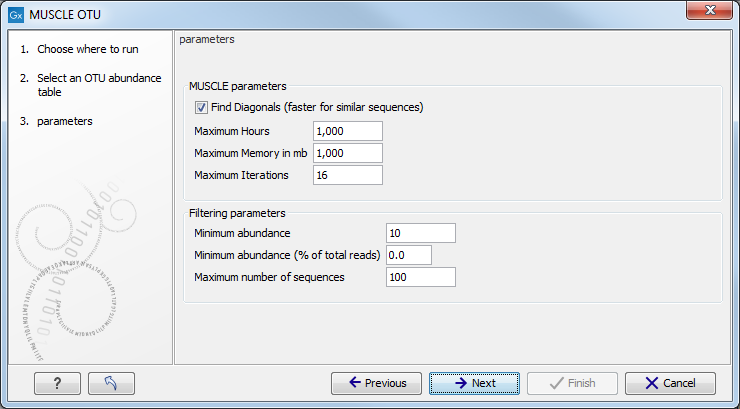
Figure 5.12:
Set up parameters for aligning sequences with MUSCLE.
- Find Diagonals: you can decide on some restrictive parameters for your analysis: the Maximum Hours the analysis should last, the Maximum Memory in mb that should be used for the analysis, or the Maximum Iterations the analysis should make. The latter is set to 16 by default.
- Filtering Parameters: The algorithm filters out all OTUs whose combined abundance across all samples is less than the minimum combined abundance or whose combined abundance is less than the minimum combined abundance (% of all the reads) across all samples. The default value for the Minimum combined abundance is set at 10. Moreover, you can specify the Maximum number of sequences to be aligned, so that only the sequences with the highest combined abundances will be used. Note that reducing the number of sequences will speed up the alignment and the construction of phylogeny trees.
Note that by default only the top 100 most abundant OTUs are aligned using MUSCLE and used to reconstruct the phylogeny tree in the next step. This phylogenetic tree is used for calculating the phylogenetic diversity and the UniFrac distances, so these measures disregard the low abundance OTUs by default. If more OTUs are to be included, the default settings for the MUSCLE alignment need to be changed accordingly.
For further analysis with the Alpha and Beta diversity tools, save the alignment and construct a phylogenetic tree using the Maximum Likelihood Phylogeny tool, available at:
Tools | Classical Sequence Analysis (![]() ) | Alignments and Trees (
) | Alignments and Trees (![]() )| Maximum Likelihood Phylogeny (
)| Maximum Likelihood Phylogeny (![]() )
)
For more information, see https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Maximum_Likelihood_Phylogeny.html.