Length trimming
Clicking Next will allow you to specify length trimming as shown in figure 21.13.
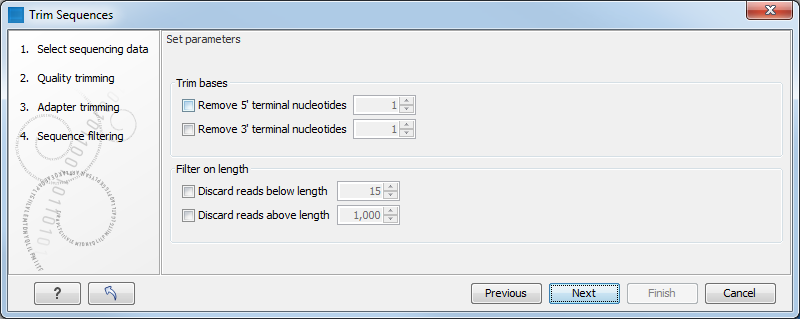
Figure 21.13: Trimming on length.
At the top you can choose to Trim bases by specifying a number of bases to be removed from either the 3' or the 5' end of the reads. Below you can choose to Discard reads below length. This can be used if you wish to simply discard reads because they are too short. Similarly, you can discard reads above a certain length. This will typically be useful when investigating e.g. small RNAs (note that this is an integral part of the small RNA analysis together with adapter trimming).