Variant tracks
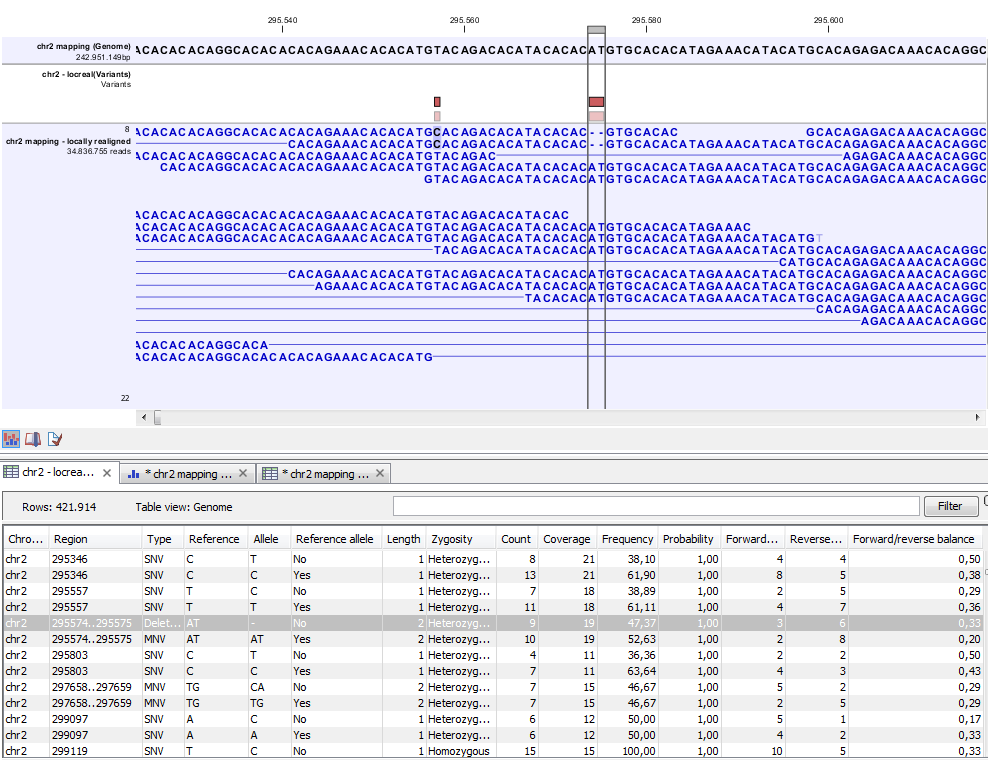
Figure 21.79: Variant track. The figure shows a track list (top), consisting of a reference sequence track, a variant track and a read mapping. The variant track was produced by running the Probabilistic Variant Caller on the read track. The variant track has been opened in a separate table view by double-clicking on it in the track list. By selecting a row in the variant track table, the track list view is centered on the corresponding variant.
A variant track (figure 21.79), created with the CLC Cancer Research Workbench variant callers (see Variant Detectors - Overview), has the following information for each variant:
- Chromosome
- The name of the reference sequence on which the variant is located.
- Region
- The region on the reference sequence at which the variant is located. The region may be either a 'single position', a 'region' or a 'between position region'. Examples are given in figure 21.80. An extract of a gvf-file giving rise to these three variants after import is shown in figure 21.81.
- Variant type
- The type of variant. This can either be SNV (single-nucleotide variant), MNV (multi-nucleotide variant), insertion, deletion, or replacement. Learn more in Variant types.
- Reference
- The reference sequence at the position of the variant.
- Allele
- The allele sequence of the variant.
- Reference allele
- Describes whether the variant is identical to the reference. This will be the case one of the alleles for most, but not all, detected heterozygous variants (e.g. the variant caller might detect two variants, A and G, at a given position in which the reference is 'A'. In this case the variant corresponding to allele 'A' will have 'Yes' in the 'reference allele' column entry, and the variant corresponding to allele 'G' would have 'No'. Had the variant caller called the two variants 'C' and 'G' at the position, both would have had 'No' in the 'Reference allele' column).
- Length
- The length of the variant. The length is 1 for SNVs, and for MNVs it is the number of allele or reference bases (which will always be the same). For deletions, it is the length of the deleted sequence, and for insertions it is the length of the inserted sequence. For replacements, both the length of the replaced reference sequence and the length of the inserted sequence are considered, and the longest of those two is reported.
- Zygosity
- The zygosity of the variant called, as determined by the variant caller. This will be either 'Homozygous', where there is only one variant called at that position or 'Heterozygous' where more than one variant was called at that position.
- Count
- The number of 'countable' reads supporting the allele. The 'countable' reads are those that are used by the variant caller when calling the variant. Which reads are 'countable' depends on the user settings when the variant calling is performed - if e.g. the user has chosen 'Ignore broken pairs', reads belonging to broken pairs are not 'countable'.
- Coverage
- The read coverage at this position. Only 'countable' reads are considered (see under 'Count' above for an explanation of 'countable' reads. Also see Detailed information about overlapping paired reads for how overlapping paired reads are treated.)
- Frequency
- The number of 'countable' reads supporting the allele divided by the number of 'countable' reads covering the position of the variant ('see under 'Count' above for an explanation of 'countable' reads).
- Probability
- The probability that this particular variant exists in the sample. (For further information please refer to the White paper on Probabilistic Variant Caller: http://www.clcbio.com/files/whitepapers/whitepaper-probabilistic-variant-caller-1.pdf).
- Forward read count
- The number of 'countable' forward reads supporting the allele (see under 'Count' above for an explanation of 'countable' reads). Also see more information about Detailed information about overlapping paired reads.
- Reverse read count
- The number of 'countable' reverse reads supporting the allele (see under 'Count' above for an explanation of 'countable' reads). Also see more information about Detailed information about overlapping paired reads.
- Forward/reverse balance
- The minimum of the fraction of 'countable' forward reads and 'countable' reverse reads carrying the variant among all 'countable' reads carrying the variant (see under 'Count' above for an explanation of 'countable' reads).21.1
- Average quality
- The average read quality score of the bases supporting a variant.If there are no values in this column, it is probably because the sequencing data was imported without quality scores (learn more about importing quality scores from different sequencing platforms in Import high-throughput sequencing data). For deletions, the quality scores of the two surrounding bases are taken into account, and the lowest value of these two is reported.
- Hyper-allelic
- Relevant for "Quality-based Variant Detection". Reports hyper-allelic status of variants based on the specified threshold "Maximum expected allele" in the "Set genome information" wizard under "Ploidy". The output in the table is "Yes" or "No" with respect to whether the threshold has been exceeded.
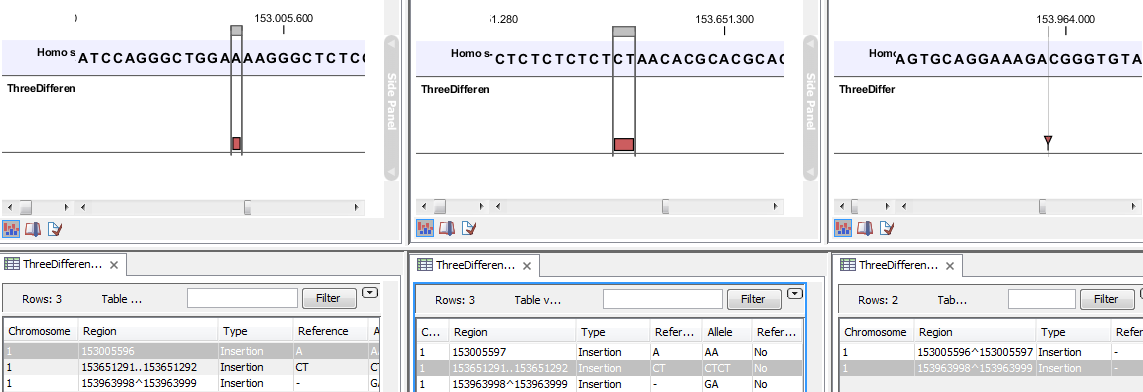
Figure 21.80: Examples of variants with different types of 'Region' column contents. The left-most variant has a 'single position' region, the middle variant has a 'region' region and the right-most has a 'between positions' region.
Please note that the variants in the variant track can be enriched with information using the annotation tools in Add Information to Variants variants.
A variant track can be imported and exported in VCF or GVF formats. An example of the gvf-file giving rise to the variants shown in figure 21.80 is given in figure 21.81.
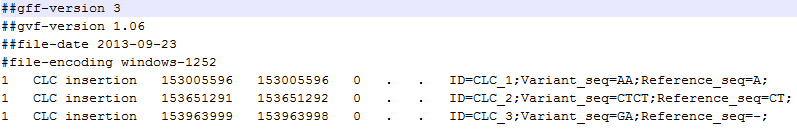
Figure 21.81: A gvf file giving rise to the variants in the figure above.
Footnotes
- ... reads).21.1
- Some systematic sequencing errors can be triggered by a certain combination of bases. This means that sequencing one strand may lead to sequencing errors that are not seen when sequencing the other strand (see [Nguyen et al., 2011] for a recent study with Illumina data). In order to evaluate whether the distribution of forward and reverse reads is approximately random, this value is calculated as the minimum of the number of forward reads divided by the total number of reads and the number of reverse reads divided by the total number of reads supporting the variant. An equal distribution of forward and reverse reads for a given allele would give a value of 0.5. (See also more information about Detailed information about overlapping paired reads.)