Trim primers of mapped reads
The Trim primers of mapped reads tool has been developed to targeted amplicon sequencing experiments with many targets (and as a consequence many primers). The tool makes use of the primer pairs in the trimming process, which greatly speeds up the trimming process time.
Removal of primers from the mapped reads ensures that no bias is introduced in the variant calling as would be the case if the primers were considered to be part of the sequencing reads. To be able to trim off the primers used in your sequencing experiment you must know the primer sequences as you will need to specify which target primer sequence file to use.
The Trim Primers of Mapped Reads can be found in the toolbox:
Toolbox | Resequencing Analysis (![]() ) | Trim Primers of Mapped Reads (
) | Trim Primers of Mapped Reads (![]() )
)
This will open the wizard shown in figure 21.6. In the first wizard step you are asked to select the read mapping. If you would like to analyze more than one read mapping, you can choose to run the analysis in batch mode by ticking the "Batch" box in the lower left corner of the wizard and then selecting the folder that hold the read mappings you want to analyze.
Figure 21.6: Select files to import.
Click on the button labeled Next to go to the next wizard step (see figure 21.7).
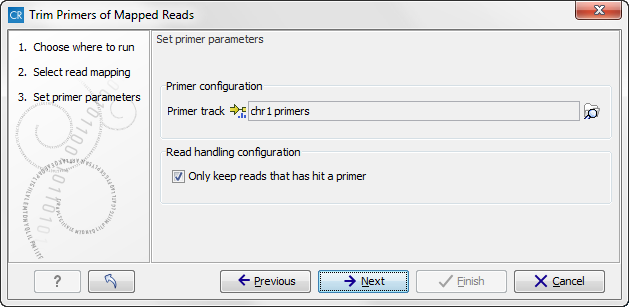
Figure 21.7: Select your primer location file and choose whether you want to keep or discard reads with no matching primers.
- Primer track Click on the folder icon in the right side to select your primer location file.
- Read handling configuration If you tick "Only keep reads that have hit a primer", reads with no matching primers will be discarded.
Click on the button labeled Next to go to the next wizard step, choose to save the result of the primer trimming and click on the button labeled Finish. The output corresponds to the input with the only difference that the primers have been trimmed off and that the output file has "trimmed reads" appended to the name.